6. Fingerprints - Screening and Similarity
Similarity measures, calculations that quantify the similarity of two molecules, and screening, a way of rapidly eliminating molecules as candidates in a substructure search, are both processes that use fingerprints. Fingerprints are a very abstract representation of certain structural features of a molecule; before we describe them, we'll discuss the problems that inspired the development of the fingerprinting techniques used in the Daylight Chemical Information System. To begin, let us define a few terms:
- pattern
- The thing for which you are searching; also called a target or substructure.
- molecule
- The thing being searched. Sometimes called the object or structure.
- N
- The "size" of a pattern. Roughly speaking, equal to the number of atoms and bonds the pattern contains (its exact definition isn't important, only the general idea).
- P «in» M
- We use the «in» symbol to mean "is a substructure of" - i.e. "P «in» M" means "pattern P is a substructure of molecule M". "P «not in» M" means "pattern P is not a substructure of molecule M".
- O(f(N))
- The order (computation time, or "cost") of performing a search. For example, O(N2) (pronounced "order N squared") means that the running time is proportional to the square of a pattern's size.
Substructure searching is known to be in the non-polynomial-complete (NP-complete) class of computational problems. Non polynomial means that the worst-case time to solve such a problem can never be expressed as a polynomial in which the number of atoms and/or bonds is the independent variable; i.e. it can't be of the form O(NK). Instead, the worst-case time for a substructure search is always of the form O(KkN).
This is quite unfortunate. To see why, imagine (for simplicity) that a substructure-search program takes 2N/106 seconds to compute where N is the number of atoms. This program can solve a 1-atom problem in a microsecond, and a 10-atom problem takes about a millisecond. This does not seem too bad until we realize that each time we add an atom we double our time: a 20-atom problem takes about a second, and a 30-atom problem takes 17 minutes. Clearly this algorithm will be inadequate as we attempt to solve real chemical problems. By contrast, if we could find an algorithm that ran in N2/103 seconds, it would be 1000 times slower on the 1-atom problem but could solve the 30-atom problem in less than a second. Clearly a polynomial solution is better than an exponential solution.
Luckily, although substructure searching is exponential in the worst case, real chemicals don't exhibit the high connectivity which, in a generalized mathematical graph, leads to worst-case behavior. Typical chemical substructure searches take O(N2) or O(N3) time, and although this is not exactly blazingly fast, it is a lot better than exponential behavior. But even this polynomial performance is slow - it can take a significant fraction of a second for a substructure search - and NP-complete theory tells us that we might occasionally run into the worst-case, an exponential-time search.
One of the cold, hard facts about NP-complete problems is that there is no way around them. If you think you've found a substructure-search algorithm that always runs in polynomial time, you should try your hand at a perpetual-motion machine. Your algorithm might work for most cases, but if it always finishes in polynomial time some of its answers must be wrong.
Fortunately there is a "hole" of sorts in these cold, hard facts: we can't detect the presence of a substructure in polynomial time, but we can often detect the absence of a substructure much faster, often linear time: O(N). The trick is to use an "imperfect" algorithm, one that can say P «not in» M with 100% confidence but that can only say P «in» M with lower confidence Such an algorithm, called a screen, does not violate any mathematical laws - ultimately we still have to use a real substructure search to get a 100%-confident P «in» M answer - but our "cheap" algorithm screens out most cases, avoiding the "expensive" algorithm most of the time.
6.1 A Brief History of Screening Large Databases
Many substructure-searching problems call for repeatedly examining a large number of molecules (typically stored in a database), comparing each with a pattern. In such situations, it pays to spend some time "up front," storing the answers to specific questions for each structure in the database. Subsequent searches of the database use these pre-computed answers to vastly improve search time; the up-front computation time is paid back quickly as repeated searches are performed.
For example, one simple screen notes the molecular formula (MF) of each molecule as it is added to the database. When a pattern is presented for searching, we generate its molecular formula; during the search, we compare the pattern's MF to each molecule's MF, and reject any molecules that are missing atoms the pattern requires. By doing this, we eliminate expensive substructure searches that are doomed to fail for the "obvious" reason of not having enough of a particular element. If the MF screen says P «not in» M, we can be 100% confident that it is correct; if the molecular formulas are compatible we have to continue with other screens or with the substructure search itself.
Molecular Formula is only one of many screens we can apply, but it illustrates the fundamental concept of screening: We only do the "expensive" substructure search when no screen can say P «in» M. By devising clever screens, we can increase the reliability of the screens to where they reject almost all structures except those that ultimately pass the substructure test (that is, the screens have very few "false positives").
6.1.1 Structural keys
Structural keys were the first type of screen employed for high-speed screening of chemical databases. A structural key is usually represented as a boolean array, an array in which each element is TRUE or FALSE. Boolean arrays in turn are usually represented as bitmaps, an array of bytes or words in which each bit represents one position of the boolean array. As the name implies, a structural key is a bitmap in which each bit represents the presence (TRUE) or absence (FALSE) of a specific structural feature (pattern).
To make a structural key, one decides which structural features (patterns) are important, assigns a bit of the bitmap to each, then generates a bitmap for each molecule in the database. Generating a structural key is time-consuming: you have to do a substructure search for each pattern represented in the bitmap, and repeat this for each molecule in the database.
The list of patterns that one might use is long. Some examples are:
- The presence/absence of each element, or if an element is common (nitrogen, for example), several bits might represent "at least 1 N", "at least 2 N", "at least 4 N", and so forth.
- Unusual or important electronic configurations, such as "sp3 carbon" or "triple-bonded nitrogen."
- Rings and ring systems, such as cyclohexane, pyridine, or napthalene.
- Common functional groups, such as alcohols, amines, hydrocarbons, and so forth.
- Functional groups of special importance in a particular database. For example, a database of organo-metallic molecules might have bits assigned for metal-containing functional groups; in a drug database one might have bits for specific skeletal features such as steroids and barbiturates.
- "Disjunctions" of unusual features. There might be patterns that are particularly rare, thus not individually worth the "cost" of a bit, yet extremely significant when they do occur. Several such patterns can be assigned to the same bit; if any one of the patterns is present the bit is set.
When a database is to be searched for a particular pattern, a structural key is generated for the pattern. As the search proceeds, the pattern's structural key is compared to that of each molecule in the database. If any TRUE bit in the pattern's key is not also TRUE in the molecule's key, then the feature represented by that bit is not in the molecule, and the pattern couldn't possibly be a substructure of the molecule. Structural keys make a very fast screen for substructure searching since computers perform the necessary boolean operations very quickly.
Structural keys vary widely in size, from a few tens or hundreds of bits to several thousand bits (in a single database, structural keys are usually all the same size since they all must represent the same thing). The choice of size is a tradeoff between specificity and space: The more bits there are, the better the chances that the screen can say P «in» M and thus avoid a full substructure search.
6.1.2 Fingerprints
The next evolutionary step in high-speed structural screening was the fingerprint, a more abstract relative of the structural key.
The structural keys described above suffer from a lack of generality. The choice of patterns included in the key has a critical effect on the search speed across the database: An effective choice will screen out virtually all structures that aren't of interest, greatly increasing search speed, whereas a poor choice will cause many "false hits," which slows searching to a crawl. The choice of patterns also depends on the nature of the queries to be made: A structural key used by a group of pharmaceutical researchers might be nearly worthless to a group of petrochemical researchers.
Fingerprints address this lack of generality by eliminating the idea of pre-defined patterns. A fingerprint is a boolean array, or bitmap, but unlike a structural key there is no assigned meaning to each bit. Your own fingerprint is very characteristic of you, yet there is no meaning to any particular feature. Similarly, a pattern's fingerprint characterizes the pattern, but the meaning of any particular bit is not well defined.
Unlike a structural key with its pre-defined patterns, the patterns for a molecule's fingerprint are generated from the molecule itself. The fingerprinting algorithm examines the molecule and generates the following:
- a pattern for each atom
- a pattern representing each atom and its nearest neighbors (plus the bonds that join them)
- a pattern representing each group of atoms and bonds connected by paths up to 2 bonds long
- ... atoms and bonds connected by paths up to 3 bonds long
- ... continuing, with paths up to 4, 5, 6, and 7 bonds long.
For example, the molecule OC=CN would generate the following patterns:
| 0-bond paths: | C | O | N |
| 1-bond paths: | OC | C=C | CN |
| 2-bond paths: | OC=C | C=CN | |
| 3-bond paths: | OC=CN | ||
The list of patterns produced is exhaustive: Every pattern in the molecule, up to the pathlength limit, is generated. For all practical purposes, the number of patterns one might encounter by this exhaustive search is infinite, but the number produced for any particular molecule can be easily handled by a computer.
Because there is no pre-defined set of patterns, and because the number of possible patterns is so huge, it is not possible to assign a particular bit to each pattern as we did with structural keys. Instead, each pattern serves as a seed to a pseudo-random number generator (it is "hashed"), the output of which is a set of bits (typically 4 or 5 bits per pattern); the set of bits thus produced is added (with a logical OR) to the fingerprint.
Note that because each set of bits is produced by a pseudo-random generator, it is likely that sets will overlap. For example, suppose we are in the middle of generating a fingerprint, and it happens that 1/4 of the bits are already set. If the next pattern generates a set containing 5 bits, the probability that all 5 bits will be unique is (3/4)5, or about 24%. Likewise, the probability that all 5 bits will not be unique are (1/4)5, or about 0.1%.
In spite of the difference between the meaning of a fingerprint's bits and a structural key's bits, fingerprints share an important feature with structural keys: If a pattern is a substructure of a molecule, every bit that is set in the pattern's fingerprint will be set in the molecule's fingerprint. This means that, like structural keys, we can use simple boolean operations on fingerprints to screen molecules as we search a database, making a fingerprint comparison an extremely fast screen for substructure searching.
The best way to think of the bits of a fingerprint is as "shared" among an unknown but very large number of patterns. Each pattern generates its particular set of bits; so long as at least one of those bits is unique (not shared with any other pattern present in the molecule), we can tell if the pattern is present or not. A structural key indicates with certainty that a particular pattern is present or absent. Fingerprints are not so definite: if a fingerprint indicates a pattern is missing then it certainly is, but it can only indicate a pattern's presence with some probability. Although a fingerprint doesn't indicate with 100% certainty that a particular pattern is present, it contains far more patterns total than a structural key, the net result being that a fingerprint is a far better screen than a structural key in almost all situations.
Fingerprints have several advantages over structural keys:
- Since fingerprints have no pre-defined set of patterns, one fingerprinting system serves all databases and all types of queries.
- More effective use is made of the bitmap. Structural keys are usually very "sparse" (mostly zeros) since a typical molecule has very few of the patterns that the structural key's bits represent. Although a mathematical analysis of fingerprint density is beyond the scope of this introduction, it turns out that fingerprints can be relatively "dense" (20-40% ones) without losing specificity. The result is that a fingerprint can be much smaller than a structural key with the same discriminating power.
- The patterns that go into a fingerprint are highly overlapped - except for "lone atoms", each pattern shares portions of itself with at least one other pattern (the example above illustrates this). The result is that the more complex a molecule gets, the more accurately its fingerprint characterizes it.
6.1.3 Variable-sized Fingerprints
The next evolutionary step in screening was the concept of folding a fingerprint to increase information density.
In the discussion above we mentioned the sparseness of a fingerprint, which is directly related to its information density. A fingerprint's information density can be thought of as the ratio how much information it actually holds to how much it could hold. As a practical definition, we measure the bit density, the ratio of "on" bits to the total number of bits (e.g. the bit density of "11000000" is 0.25).
Fingerprints for small molecules and "featureless" molecules (such as CH4 or C40H82) have less information in them than those for large or "rich" molecules. But the fingerprinting mechanisms discussed so far require a fixed fingerprint size for all molecules. If we choose to use small fingerprints, the fingerprint of large or complex molecules will be "black" - nearly all ones - and will not discriminate well (there is more information than the fingerprint can hold). On the other hand, if we use very large fingerprints, most molecules' fingerprints will be "white" - nearly all zeros - and will waste space. In both cases we have low information density; the "black" fingerprint because it is too dense and the "white" one because it is too sparse.
Ideally, we would like to choose a particular discriminatory power (e.g. "For a typical pattern, 98% of the database is screened out by the pattern's fingerprint") and compute the fingerprint density needed to achieve that discriminatory power on a case-by-case basis. Although we can not actually do this, the process of "folding" achieves nearly the same performance and size as the "ideal" case.
The folding process begins with a fixed fingerprint size that is quite large - large enough to accurately represent any molecule we expect to encounter. The fingerprint is then folded: we divide it into two equal halves then combine the two halves using a logical OR. The result is a shorter fingerprint with a higher bit density. We can repeatedly fold the fingerprint until the desired information density (called the minimum density) is reached or exceeded.
As long as two fingerprints are the same size (even if created with different sizes), they are compatible. To see why, consider the fingerprints of a pattern P and a molecule M. If the screen is initially positive (e.g. all bits in the P's fingerprints are also in M's) then the same will be true after folding. On the other hand, a negative screen (at least one bit in P's fingerprint is not in M's) might be converted to a positive screen after folding. But this is ok - converting "correct negative" to a "false positive" doesn't violate the rules of screening: a screen is only required to say P «in» M with 100% reliability. With each fold, we increase the chances of a false positive but save half of the space needed to store the fingerprint.
Fingerprint folding allows us to optimize the information density in a set of fingerprints, thus optimizing screening speed. Rather than choosing one fingerprint size for the entire database, we choose the size of each molecule's fingerprint individually, according to the complexity of the molecule and the desired success rate of the screening process. In most real databases, optimizing the information density greatly reduces the amount of data stored, and increases the screening speed correspondingly.
6.1.4 In-memory Screening
Although there is no reason a structural key can't reside in memory rather than on disk, they tend to be considerably larger than fingerprints, too large to fit a reasonable number of them into memory. This was especially true when structural keys were first developed - memory was several orders of magnitude more expensive than it is now, and most computers had precious little of it. Thus, structural-key screening has almost always been done as a disk-based screening technique.
Computer memories are roughly 105 times faster than disks. If one can read the bitmaps of a structural key or fingerprint into memory and search it there, the speed of the screen itself increases a corresponding amount. About the time fingerprints were developed, computer memory prices reached a point where the fingerprints from a relatively large database all could be loaded into memory.
With the advent of in-memory fingerprint-based screening, exploratory data analysis takes on a completely new aspect. One can quite literally "explore" a database, trying different searches, refining them, taking side-tracks to see where they would lead, and so on.
The evolution of the above-described screening techniques has now reached the point where the SMILES and fingerprints of all chemicals known in the world (roughly 10-15 million structures) can fit in the memory of a large workstation-class computer; it doesn't even take a "mainframe" computer, much less a supercomputer. An ordinary database of tens or hundreds of thousands of molecules can easily fit into the memory of today's run-of-the-mill workstations. Speed increases are correspondingly dramatic: an ordinary workstation can screen 100,000 to 1,000,000 structures per second using in-memory folded fingerprints.
The number of molecules that can be considered in a single search has also grown dramatically. It is now actually possible to search all known chemicals in the world in a reasonable time (that is, if you can get your hands on such a database). Most users, using a corporate or academic database of less than a million molecules, will be able to search their database in a few seconds - the time it takes to answer the question is now much shorter than the time it takes to think of the question, even on relatively large databases.
6.2 Fingerprints and Reactions
There are two distinct types of fingerprints which the Daylight system provides for reaction processing. First is the "normal" structural fingerprint, which is useful as a superstructure search screen. Second is the "difference" fingerprint, which is useful as a metric for the bond changes which occur during a reaction.
6.2.1 Structural Reaction Fingerprints
The structural reaction fingerprint is nothing more than the combination of the normal structural fingerprints for the reactant and product molecules within the reaction. The combination is the bitwise-OR of the following fingerprints:
- Fingerprint of the reactant part
- Fingerprint of the product part
- Bit-shifted fingerprint of the product part
Once the structural reaction fingerprint has been generated, all of the normal fingerprint operations (folding, similarity, substructure screen) apply.
6.2.2 Reaction Difference Fingerprints
The difference fingerprint is a new type of fingerprint specifically tailored for reaction processing. If one has a reaction which is stoichiometric (all atoms on the reactant side appear on the product side), then the difference in the fingerprint of the reactant molecules and the fingerprint of the product molecules will reflect the bond changes which occur during the reaction.
On the surface, one could perform an "exclusive-OR" operation between the reactant and product fingerprint. Unfortunately, since the molecule fingerprints do not keep track of the count of paths many of the relevant bonds get masked out during the "XOR" operation. What is required is to keep track of the count of each path in the reactant and product and then subtract the counts of a given path. If the difference in count is non-zero, then the path is used to set a bit in the difference fingerprint. If the difference in count is zero, then no bit is set for that path in the difference fingerprint.
For example, consider our well-worn Sn2 displacement reaction:
The paths generated for the molecules would be as follows:
| Enumerated Fingerprint Paths: | ||
| Path Length: | Reactant (count/path): | Product (count/path): |
| 0 | 1 I, 1 Na, 3 C, 1 Br | 1 I, 1 Na, 3 C, 1 Br |
| 1 | 1 C=C, 1 C-C, 1 C-Br | 1 C=C, 1 C-C, 1 C-I |
| 2 | 1 C=C-C, 1 C-C-Br | 1 C=C-C, 1 C-C-I |
| 3 | 1 C=C-C-Br | 1 C=C-C-I |
| Difference in Path Counts: | |
| Path Length: | Difference (count/path): |
| 0 | 0 I, 0 Na, 0 C, 0 Br |
| 1 | 0 C=C, 0 C-C, 1 C-Br, 1 C-I |
| 2 | 0 C=C-C, 1 C-C-Br, 1 C-C-I |
| 3 | 1 C=C-C-Br, 1 C=C-C-I |
After generating the difference in counts, we only use the six paths with non-zero differences to set bits in the difference fingerprint. These are the paths which walk through bonds that change during the reaction. By considering only these paths, we get a fingerprint which reflects the overall bond changes in the reaction.
Once generated, the difference fingerprint is a normal fingerprint and can be folded, used for similarity measurements and clustering. The difference fingerprint may not be used as a substructure screen for any types of searching, since it does not obey the strict subset relationship required for screening. In the above example, note that the "[Na+]" atom does not appear in any of the difference fingerprint paths. Thus, any search query which contained Sodium-containing paths would not pass the screening step, even if a valid query match.
6.3 Similarity Measures
One often wishes to know how similar one molecule is to another, independent of whether either is a substructure of the other. There are many ways one might choose to measure such similarity; for example a chemical approach might rank them according to the number of physical properties and reactions they share, whereas a mathematical approach might rank them according to their similarity in three dimensions.
The Daylight fingerprints described above effectively encode the substructures present in a molecule. It would not seem unreasonable that the proportion of substructures in common between two molecules should be a reasonable measure of similarity of the overall molecules. In mathematical terms this is a comparison of the bits in the fingerprints which are set on.
Note that the similarity measures are independent of the molecular feature descriptors. Fingerprints generated from structural keys or any other method can be used in the Daylight toolkits. The only requirements are that the size is a power of 2 and a multiple of 8. This ensures folding and translation to ascii representation works correctly. The current size limitations are 32 to 230
If we describe our molecules by the presence or absence of features, then the binary association coefficients or similarity measures are based on the four terms a, b, c, d shown in the two way table.
| OBJECT B | ||||
|---|---|---|---|---|
| 0 | 1 | Totals | ||
| OBJECT A | 0 | d | b | b+ d |
| 1 | a | c | a + c = A | |
| Totals | a + d | b + c = B | n | |
Where:
a is the count of bits on in object A but not in object B.
b is the count of bits on in object B but not in object A.
c is the count of the bits on in both object A and object B.
d is the count of the bits off in both object A and object B.
In addition:
n = ( a + b + c + d )
A = ( a + c )
B = ( b + c )
Where:
n is the total number of bits on or off in objects A
or B.
A is the count of the bits on in object A.
B is the count of the bits on in object B.
N.B. This nomenclature differs from that used by others, in particular the Sheffield group. In their system the labels a and c are reversed.
Pre 4.5 releases of the Daylight software provided an example of each of
these classes.
- The Euclidian coefficient, E is defined as the square root of
the ratio:
( c + d )/n As both numerator and denominator are functions of d, E belongs to the first class of association coefficients dependent on the double zeros.
As we do not take the square root, given mostly we are only interested in ranking ; it is simply a matching coefficient (Sokal, R.R., Michener, C.D., (1958) The University of Kansas Scientific Bulletin 38, 1409-1438). In reality the value returned for this coefficient is the complement of this, i.e. the Hamming distance, or the Total Difference Coefficient (Sneath, P.H.A., (1968) Journal of General Microbiology 54, 1-11)( a + b )/n
- The Tanimoto coefficient, T, is defined as the ratio:
c/(a + b + c) T is independent of d, i.e. T belongs to the second class of association coefficients defined above. It may be regarded as the proportion of the "on-bits" which are shared. See Tanimoto, T.T. (1957) IBM Internal Report 17th Nov see also Jaccard, P. (1901) Bulletin del la Société Vaudoisedes Sciences Naturelles 37, 241-272 .
N.B. These association coefficients are not necessarily true metrics. In particular those where the divisor depends on the particular pairwise comparison i.e. is not equal to n, may violate the triangle inequality. See Anderberg, MR (1973) Cluster Analysis For Applications, Academic Press p 117, for further discussion.
Over the years there has been much discussion as to which type of coefficient to use. In chemistry it has generally been thought that, as most descriptor features are absent in most molecules, i.e. the bit string descriptors such as the Daylight fingerprint contains mainly zeros, coefficients such as the Tanimoto are more appropriate. Further, given that the size of a Daylight fingerprint can be arbitrarily doubled, thereby adding mainly random off bits, any measure using matching off-bits, d, would be inappropriate. However this may not be the case for fixed width key based fingerprints. Daylight therefore offers access to both types of measure. The user must ensure that an appropriate one is chosen.
In version 4.5 Daylight extended the range of coefficients which could be used by introducing the asymmetric Tversky index. This allowed users to make use of directional similarity and harness the power of the concepts of prototypes in similarity searching.
As has been indicated above all of these indices are not monotonic, and as early as 1982 Hubalek (see Hubalek, Z. Biol. Rev. (1982) 57, 669-689) showed that the coefficients could be clustered on the ranking of a given set of objects.
Recently Holliday et al ( Holliday, JD., Hu, C-Y. and Willett, P. (2002) Combinatorial Chemistry and High Throughput Screening 5, 155-166 ) have shown that a whole range of similarity measures can be clustered on the ranking of chemical structures.
With the release of version 4.9 therefore, we have introduced a whole range of named similarity measures and additionally allowed users to construct their own, from the terms a, b, c, d as appropriate.
6.3.1 Tversky Index
As of Release 4.51 the Tversky index (Tversky, A. Psychological
Reviews (1977)84 (4) 327-352), provides a more powerful method for
structural similarity searching. Its use and interpretation, however, are
not not as simple as the Tanimoto index. The Tversky comparison is
intrinsically asymmetric. As with Tanimoto the features present in two
objects are compared. In the Tversky approach we have the concept of a "prototype"
to which the objects or "variants" are compared. Note
this differs from the Tanimoto index in which the similarity between
two objects is estimated. This inherent asymmetry means that the Tversky
index is very definitely not a metric. The ratio model in which
the value is bounded (between 0 and 1) is defined as follows:
c/(α* a + β* b + c)
Setting the weighting of prototype features to the same value does not use the power of this index. Indeed, setting α = β= 1, produces the Tanimoto index. If α = β= 0.5 we get the Dice index. See below. The value of the index comes from setting the weighting of prototype and variant features asymmetrically, producing a similarity measure in a more-substructural or more-superstructural sense or reflecting the increased knowledge the user has about the prototype. Quite often one is looking for compounds like a known compound with appropriate properties.
Setting the weighting of prototype features to 100% (α=1) and variant features to 0% (β=0) means that only the prototype features are important, i.e., this produces a "superstucture-likeness" measure. In this case, a Tversky similarity value of 1.0 means that all prototype features are represented in the variant, 0.0 that none are.
Conversely, setting the weights to 0% prototype (α=0) / 100% variant (β=1) produces a "substructure-likeness" measure, where completely embedded structures have a 1.0 value and "near-substructures" have values near 1.0. Note: with no weight at all given to variant features, this measure is pretty sensitive to "noise" in Daylight fingerprints and settings of 90%/10% generally produce a more useful ranking.
Tversky measures where the two weightings add up to 100% (1.0) are of special interest. In XVMerlin the Tversky search query panel provides a "Sum 100%" checkbox which, when selected, forces the two weights to add up to 100%.
Advanced users may wish to experiment with Tversky indices where weightings are not limited to 100%. Weightings greater than 100% causes the distinguishing features a, b to be emphasized more than common features, c, which may be useful in analysis of diversity or dissimilarity.
6.3.2 User-defined and Named Similarity indexes
With the 4.9 release, users are able to use their own similarity measure throughout the software, where before they have been restricted to the hardcoded measures described above. The measure is entered as a string representing a f( a, b, c, d) with all of the usual mathematical expressions available. There is no restriction on the form of the function, but to work as a measure of similarity it should fulfill certain requirements. Expressions such as:
(c + d) /( a + b + c )
2.0*(c +d)/( a + b + 2.0*c )
(c + d)/( 2.0* ( a + b ) + c )
are not useful, even though the user has attempted to alter the weights of the matched/unmatched pairs. It is nonsense to include in the numerator that which has been specifically excluded in the denominator (Anderberg, MR (1973) Cluster Analysis For Applications, Academic Press p. 89).
Users should also be cognizant of the restrictions imposed by the descriptors being compared. Daylight fingerprints can be arbitrarily doubled in size, as described above. The value of d, can be increased enormously without scaled increases in the information rich on-bits. It is not recommended therefore that indexes based on a function which uses the value of d, are used with Daylight type fingerprints. In the case of fingerprints based on structure keys there may be no such restrictions.
When doing comparisons users should also be aware of the range of possible values the index can take. Most have the range 0.0, 1.0 but this is not mandatory.
The most common useful indexes have been collected by Holliday et al (Holliday, JD., Hu, C-Y. and Willett, P. (2002) Combinatorial Chemistry and High Throughput Screening 5, 155-166) These are shown in the table, and can be referred to, by name, in applications and toolkits calls which allow user defined similarity functions.
| Measure | Range | Formula |
| Cosine | 0.0,1.0 | 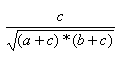 |
| Dice | 0.0,1.0 | 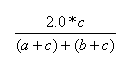 |
| Euclid | 0.0,1.0 | 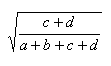 |
| Forbes | 0.0,∞ | 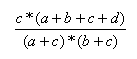 |
| Hamman | -1.0,1.0 | 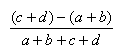 |
| Jaccard | 0.0,1.0 | |
| Kulczynski | 0.0,1.0 | 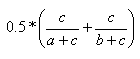 |
| Manhattan | 1.0,0.0 | 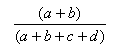 |
| Matching | 0.0,1.0 | 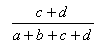 |
| Pearson | -1.0,1.0 | 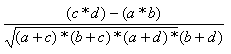 |
| Rogers-Tanimoto | 0.0,1.0 | 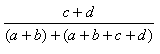 |
| Russell-Rao | 0.0,1.0 | 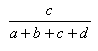 |
| Simpson | 0.0,1.0 | 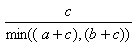 |
| Tanimoto | 0.0,1.0 | 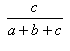 |
| Yule | -1.0,1.0 | 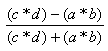 |
Notes
- The Tanimoto and Jaccard indexes are the same.
- The Forbes index has no upper limit.
- The Manhattan index is a distance = 1.0 - Matching index
- The Kulczynski index is the mean of the individual substructure similarities
- The Simpson index is the best of the individual substructure similarities
- The Dice index is the ratio of the bits in common to the arithmetic mean of the number of on bits in the two items.
- The Cosine index is the ration of the bits in common to the geometric mean of the number of on bits in the two items.
Go To Next Chapter... 7. THOR - Chemical Database System