Isotopic specifications in SMILES are indicated by prefixing the atomic symbol with a number equal to the desired integral atomic mass. An atomic mass can only be specified inside brackets.
| Depiction | SMILES | Name | Remark |
|---|---|---|---|
| C | methane | Carbon's mass is unspecified. | |
| [C] | elemental carbon | Carbon's mass is unspecified. | |
| [12C] | elemental carbon-12 | There's nothing "special" about carbon-12 just because its the most common isotope. | |
| [13C] | elemental carbon-13 | Take care, any mass can be specified, not just reasonable ones. | |
| [13CH4] | C-13 methane | Connected hydrogens must be specified inside brackets. |
Configuration around double bonds is specified in SMILES by the characters `/' and `\' which are "directional bonds" and can be thought of as kinds of single bonds. These symbols indicate relative directionality between the connected atoms and have meaning only when they occur on both atoms which are double bonded.
An important difference between SMILES chirality conventions and others such as CIP is that SMILES represents local chirality (as opposed to absolute chirality), which allows partial chirality specification.
| Depiction | SMILES | Name | Remark |
|---|---|---|---|
| F/C=C/F or F\C=C\F |
trans-difluoroethene | The F's are on "opposite sides" of the double bond. | |
| F/C=C\F or F/C=C\F |
cis-difluoroethene | The F's are on the "same side" of the double bond. | |
| F/C=C/C=C/C | trans,trans-1-fluoro-penta-1,3-diene | Double bond orientation is completely specified. | |
| F/C=C/C=CC | trans,unspec-1-fluoro-penta-1,3-diene | Double bond orientation is partially specified. |
SMILES uses a very general type of chirality specification based on local chirality and symmetry point groups. Instead of using a rule-based numbering scheme to order neighbor atoms of a chiral center, orientations are based on the order in which atoms occur in the SMILES string. As with all other aspects of SMILES, and valid order is acceptable.
The simplest and most common kind of chirality is tetrahedral: four "neighbor" atoms are evenly arranged about a central atom, known as the "chiral center". If all four neighbors differ from each other in any way, mirror images of the structure will not be identical. The two mirror images are known as "enantiomers" and are the only two forms that a single tetrahedral center can produce. If two (or more) of the neighbors are identical, the central atom will not be chiral (the mirror images can be superimposed in space).
In SMILES, tetrahedral centers may be indicated by a simplified chrial specification (@ or @@) written as an atomic property following the atomic symbol of the chiral atom. If a chiral specification is not present for a chiral atom, the chirality of that atom is implicitly not specified.
Looking at the chiral center from the direction of the "from" atom (as per
atom order in SMILES),
@ means "the other three atoms are listed anti-clockwise;
@@ means clockwise.
If all atoms are explicitly specified in SMILES,
e.g., N[C@](C)(F)C(=O)O,
the order of the atoms should be clear, i.e., N is the "from" atom,
and the other atoms are anticlockwise in
SMILES order (methyl, fluoro, carboxy):
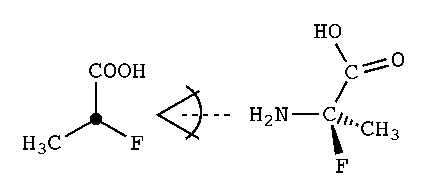 N[C@](C)(F)C(=O)O
N[C@](C)(F)C(=O)O
If the chiral atom is the very first atom in the SMILES,
e.g., [C@](F)(N)(C)CC,
the first-appearing neighbor is taken to be the "from" atom.
If the chiral atom has a non-explicit hydrogen,
(it can have at most one and still be chiral)
it will be listed inside the chiral atom's brackets,
e.g., F[C@H](N)C.
The order of the non-explicit hydrogen is exactly as written in SMILES,
i.e., in this case, the first of the three following atoms (H,N,C).
Similarly, if a chiral atom has a ringclosure, e.g., N1CCCO[C@H]1CC,
the O is the from atom, and three following atoms are in the order they
are connected to the chiral center as written in SMILES,
i.e., H (immediately following the symbol),
then N (the ring closure is next),
then the ethyl carbon.
To reiterate: the implied chiral order is always exactly as written in SMILES.
| Depiction | SMILES | Name | Remark |
|---|---|---|---|
| N[C@@H](C)C(=O)O | L-alanine | From N: (H,methyl,carboxy) appear clockwise. | |
| N[C@H](C)C(=O)O | D-alanine | From N: (H,methyl,carboxy) appear anti-clockwise. | |
| O[C@H]1CCCC[C@H]1O | cis-resorcinol | On first chiral carbon, looking from the hydroxy O: (H,CO,C) are anti-clockwise. | |
| C1C[C@H]2CCCC[C@H]2CC1 | cis-decalin | "Cis/Trans" ring fusions are really tetrahedral chiralities |
There are many kinds of chirality other then tetrahedral. The use of the @ symbol described above is actually a special case of the general SMILES chiral specification syntax:
chival : '@' <chiclass> <chiorder>
| chiral '@'
;
i.e., the chiral value is composed of the chiral class and chiral order.
The chiral class is a two-letter code named after the base
point group, e.g., tetrahedral (TH), allenyl (AL), square-planar (SQ),
trigonal-bipyramidyl (BP), octahedral (OH), etc.
The chiral order is a positive integer indicating which chiral
configuration of the given class is present
(including reduced and degenerate chiralities where the number
of distinct enantiomers is reduced by symmetry).
To simplify input of common chiralities, one chiral class is designated the default chiral class for a given degree (connectivity). For instance, the default chiral class for degree 4 is TH. Notations in the form "@@" are interpreted as @2 (analogous to "++" meaning +2). Whenever possible, the chiral order 1 ("@1" or just "@") corresponds to "anticlockwise about the the axis represented by SMILES order". ["@" is supposed to be visual mneumonic in that the symbol looks like an anticlockwise spiral about a central spot.]
The following table provides examples of a few common non-tetrahdral chiralities. More detailed information is available elsewhere.
| Class | Values | Example | SMILES | Remark |
|---|---|---|---|---|
| Allene-like | @AL1 or @AL2 | OC(Cl)=[C@]=C(C)F same as OC(Cl)=[C@AL1]=C(C)F |
Default for degree 2. As if: |
|
| Square- planar |
@SP1 to @SP3 | F[Po@SP1](Cl)(Br)I | Not a default class. @SP1 makes a "U". @SP2 makes a "4". @SP3 makes a "Z". |
|
| Trigonal- bipyramidal |
@TB1 to @TB20 | O=C[As@](F)(Cl)(Br)S | TB is the default class for degree 5. | |
| Octahedral | @OH1 to @OH30 | O=C[Co@](F)(Cl)(Br)(I)S | OH is the default class for degree 6. |