MOLSMART with Reactions
Version 2.1
A program to convert molecule or reaction queries
represented by MDL Molfiles, RGfiles and Rxnfiles
to Daylight SMARTS or SMIRKS Strings
Written by Annette von Scholley-Pfab
December 1998
Program Documentation
-
Introduction
-
Syntax and Options
-
Conversion of Atom and Bond Types and Properties
-
Conversion of Link Atoms
-
Conversion of R-groups
-
Conversion of reactions
-
Program Limitations
-
Support and Further Information
Copyright (c) Barnard Chemical
Information Ltd., 1997-8
Updated 9 December 1998
1.0 Introduction
MOLSMART converts structure or reaction queries expressed in various
MDL Information Systems Inc. file formats (such as can be created using
MDL's ISIS/Draw program) to Daylight Chemical Information Systems' SMARTS
or SMIRKS strings (which can be used for searches using Daylight's MERLIN
system).
The input formats supported are Molfiles (representing simple substructure
queries), RGfiles (which can include "R-groups" and other features) and
Rxnfiles (which describe full or generic chemical reactions). Details of
these formats are given in MDL's
own documentation.
The output format is a Daylight SMARTS pattern, which for reaction queries
(Rxnfiles) will be a Reaction SMARTS. As a user-specified option, for appropriate
Rxnfiles, MOLSMART is able to output a Daylight SMIRKS string, which
is a restricted type of Reaction SMARTS pattern describing a generic reaction.
When generating SMIRKS, MOLSMART automatically checks that the data
in the Rxnfile conforms to the requirements for SMIRKS and, as a user option,
is able to assign atom-atom map classes automatically to the atoms
involved, if these are not already specified. Details of the SMARTS and
SMIRKS languages and the restrictions applied to SMIRKS are given in Daylight's
own documentation.
Normally, a single MDL file is converted to a single SMARTS pattern,
though in some cases (e.g. where the MDL file contains R-group logic ("IF
R1 THEN R2", etc.) or information about stereochemical reactions ("inversion/retention
flag")) there may be more than one, which should be ORed together in searching.
The differences in expressive power between the two formats mean that in
a few cases a precise conversion is not possible; these are noted in Section
7.
2.0 Syntax and Options
MOLSMART is able to operate either in non-interactive mode (converting
a single MDL file) or interactive mode (converting several MDL files in
response to user prompts. In both modes the resulting SMARTS or SMIRKS
strings are written to the standard output (stdout) channel, while error
and other messages are written to the standard error (stderr) channel.
2.1 Non-interactive Mode
In this mode the program is invoked by the command line:
MOLSMART [filename] [-k] [-m]
where the three arguments to the program are all optional and have the
following meanings:
| filename |
the name of the MDL file to be converted. If omitted MOLSMART
will read the input file from the standard input (stdin) channel. Since
the output SMARTS or SMIRKS is written to stdout, this allows MOLSMART
to be used as a UNIX filter. |
| -k |
MOLSMART will generate SMIRKS instead of SMARTS. If the input
file does not conform to the requirements for SMIRKS, an error message
is written to the standard error (stderr) channel. Only atom map classes
which are specified in the input file will appear in the SMIRKS. |
| -m |
This is an extension of the -k option. SMIRKS will be generated, and
provided the input reaction is balanced, or could be balanced by the addition
of a single trivial reagent or by-product, MOLSMART will automatically
assign atom map classes to any atoms which do not have map classes
specified in the input file. |
Note that Windows versions of MOLSMART will operate in interactive
mode if no command line arguments are supplied.
2.2 Interactive Mode
In this mode the program is invoked by the command line:
The "-i" argument is not necessary for Windows versions of MOLSMART,
which automatically start in interactive mode if no command line arguments
are given. In interactive mode MOLSMART will repeatedly prompt the
user to enter the filename of an MDL file to be converted. If <cr> is
pressed in response to this prompt without typing a filename, MOLSMART
will terminate. The filename typed in response to the prompt may optionally
be followed by the -k or -m argument, which will cause generation of SMIRKS
as in non-interactive mode.
2.3 Auxiliary Files
MOLSMART uses two auxiliary files which are supplied with the program
and must reside in the directory from which it is run. These are
3. Conversion of Atom and Bond Types
and Properties
The following notes discuss various points about the conversion of atom
and bond types and properties, and their representation in SMARTS patterns.
3.1. Atom Symbols
The usual SMILES atom symbols are used. Explicit hydrogens and atoms that
may be aromatic or not are represented as
"#number", e.g. [#1] = hydrogen; [#6] = aromatic or aliphatic carbon.
The MDL atom type "Q" is converted to SMARTS "[!#6]"; if it carries
a positive charge it becomes "[!#6;*+1]".
The MDL atom type "A" is converted to "*". If aromatic, this is changed
to "a", and if certainly not aromatic, to "A".
3.2. Implicit Hydrogens
In MDL, the hydrogen count values given in the atom block are minima (e.g.
H1 means at least one implicit hydrogen), whereas in SMARTS h1 means exactly
one implicit hydrogen. Thus MDL's H2 is converted to SMARTS as ";!h0;!h1".
Implicit hydrogens within a reaction centre are converted to exact hydrogen
counts (see discussion of program limitations in section
7.4).
3.3 Ring Bond Count
The MDL ring bond count (RBD entries in Properties Block) is converted
as follows:
-
-1 (no rings) gives the SMARTS "r0"
-
> 1 gives a recursive SMARTS such as [C;$(*(@*)(@*)@*)] for an C atom with
3 ring bonds. Within MDL RBD 2 or 3 means an exact number of ring bonds.
The recursive SMARTS only expresses a minimum of ring bonds. MDL RBD 4
("at least 4 ring bonds") is translated correctly.
-
-2 (as drawn): all bonds which are ring bonds in the graph or which are
explicitly declared as ring bonds are counted together and then treated
as a normal ring bond count with this value.
e.g. (i): for two carbons connected by a single bond, with RBD = -2 for
both atoms, the following SMARTS is generated: "[C;r0][C;r0]". (Of course,
RBD = -1 produces the same result.)
e.g. (ii): applied to ring systems -2 is useless because it doesn't
limit the number of ring bonds: for cyclobutane, all atoms with RBD= -2,
the following SMARTS is generated:
[C;$(*(@*)@*)]1[C;$(*(@*)@*)][C;$(*(@*)@*)][C;$(*(@*)@*)]1
3.4. Other Atom Properties and Attributes
The MDL Substitution Count property is converted to the SMARTS "D" primitive.
The MDL atom block valence specification is converted to the SMARTS
"X" primitive.
Charges are written, for example as "+2" or "-1". If in the MDL file
a zero charge is specified it is ignored.
The MDL property "unsaturated atom" ("M UNS") is converted to a recursive
SMARTS: "$(*=,#*)".
The obsolete MDL atom list block is not interpreted but the "M ALS"
property line is transformed to expressions as [Cl,Br,F] or, if the exclusion
flag is T (indicating a NOT list), to [!Cl;!Br;!F]
3.5. Bond Types and Properties
Single, aromatic and single-or-aromatic bonds are normally not written
explicitly in the SMARTS string. Exceptions are:
-
a single bond between two aromatic atoms (e.g. "c-c" indicates two aromatic
carbons joined by an aliphatic bond)
-
bonds with chain or ring specification (e.g. "-;!@" indicates single ring
bond).
The MDL bond type "double-or-aromatic" is written as "=,:".
MOLSMART performs no automatic detection of aromatic bonds; it
transcribes whatever is specified in the MDL file, which may affect the
results obtained by using the resulting SMARTS pattern for searching.
3.6. Aromaticity
In the Daylight system aromaticity is regarded as an atom property, whilst
MDL software regards it as a bond property.
MOLSMART therefore proceeds as follows:
The following atoms are set to "aromatic":
Atoms which have at least one aromatic bond.
The following atoms are set to "aliphatic":
Atoms which are in a chain (ring bound count = -1)
Atoms which have more than one implicit hydrogen
Atoms which have more than one single bond
Atoms which have either a double bond or a triple
bond
Atoms which have a defined substitution count which
is not 2 or 3
Atoms which have a defined valence which is not
3.
Atoms other than B, C, N, O, Al, Si, P, S
All other atoms are set to "aromatic-or-aliphatic".
Note that MOLSMART carries out no automatic perception of aromaticity.
If alternating single and double bonds have been
specified in the MDL input file, these will be reproduced in the SMARTS
pattern which is generated. This may result in a failure
to match against SMILES in which aromatic atoms or bonds are shown.
It is therefore recommended that bonds in the Rxnfile
to be converted be specified as aromatic, single/aromatic or double/aromatic
where appropriate.
Automatic perception of aromatic rings is planned for future versions
of MOLSMART.
3.7 Stereo Chemistry
MDL's "atom stereo parity" is translated to SMARTS chiral specifications.
e.g. (i) D-alanine: [C@H](C(=O)[#8-1])([#6])[#7+1]
e.g. (ii) L-alanine: [C@@H](C(=O)[#8-1])([#6])[#7+1]
Cis and trans stereochemistry is recognised:
e.g. (iii) cis: 1,2 dichloroethene: C(\[Cl])=C\[Cl]
e.g. (iv) trans 1,2 dichloroethene: C(\[Cl])=C/[Cl]
4. Conversion of Link Atoms
MDL Link Atoms indicate "nose-to-tail" repetition of parts of a structure.
Two different procedures are used for converting them, depending on the
presence of ring closure bonds around them.
4.1 Link Atoms without open Ring Closure Bonds
If the Link Atom is not part of a ring, a single SMARTS string can be produced,
using a series of recursive SMARTS specifications. Thus for a structure
in which two aliphatic nitrogens are linked by a chain of 1 to 3 carbons:

the following SMARTS is produced:
N [ $(C(N) N), $(C(N) CN), $(C(N) CCN) ]
(spaces have been added here to improve clarity). Note that the initial
N is repeated as part of each alternative recursive SMARTS. This is necessary
as the SMARTS
N [ $(CN), $(CCN), $(CCCN) ]
would match any structure which just has a NC in it.: To avoid this
MOLSMART
adds to the recursive SMARTS the atom to which the Link Atom is connected
and the direct neighbours of this atom. Only normal neighbours are considered
(no R-groups or other Link Atoms), and this "constant part" always appears
as the first neighbour of the first atom described by the recursive SMARTS:
4.2 Link Atoms with open Ring Closure bonds
If the Link Atom is part of a ring, separate SMARTS are enumerated for
each alternative for the Link Atom. Thus

results in the following three SMARTS which should be searched in OR
relationship
N1 C N1
N1 CC N1
N1 CCC N1
(again spaces have been added to the SMARTS for clarity).
5. Conversion of R-groups
5.1 R-groups with a single attachment point
If an R-group has more than one alternative then each alternative with
more than one atom, is expressed as recursive SMARTS (which also includes
the connected atom from the "constant part" and its direct neighbours).
If no M LOG property line appears in the RGfile, no complications arise
and a single SMARTS is produced:
R1-C-C-R2 with R1 = Cl or NO2 and R2 = ethyl
results in:
C ( [ $(N(CC)(=O)=O), Cl ] ) C[C;D2][C;D1]
5.1.1 IF R1 THEN R2 Logic
If in the above query, the following logic is specified:
-
Occurrence > 0 for both groups
-
RestH off for both groups
-
IF R1 THEN R2
three SMARTS are produced which have to be searched in OR relationship:
-
R1 is NO2 or Cl which implies that R2 must be ethyl:
[C;D2] ( [$(N(CC)(=O)=O), Cl ] ) C [C;D2] [C;D1]
-
R1 is neither NO2 nor Cl, so R2 can be anything:
[C;D2] ( [ !$(N(CC)(=O)=O) ; !Cl ] ) C
-
R1 = hydrogen so R2 can be anything
[C;D1] C
Note: the first carbon has been given a degree attribute: [C;D2] or [C;D1]
which prohibits any further substitution on this carbon (see Section 6.4,
Limitations); in the ISIS PowerSearch module further substitution would
be allowed.
5.1.2 RestH ON
In the above query, with the following logic:
-
Occurrence > 0 for both groups
-
If R1 then R2
-
RestH on for R1
the following two SMARTS result:
-
[C;D2] ( [ $(N(CC)(=O)=O), Cl ] ) C [C;D2] [C;D1]
-
[C;D1] C
and if RestH is on for both groups:
-
[C;D2]([$(N(CC)([#8])=O),Cl])C[C;D2][C;D1]
-
[C;D1][C;D1]
5.1.3 Several Occurrences at one position
Where several occurrences are permitted at one position, some complications
arise. Consider the query CH3NR1 with R1 = phenyl, and the following
three data base structures:
CH3NHPh
CH3NPh2
(CH3)2NPh
With no logic specified, the SMARTS
is produced which matches all three structures.
If the occurrence count for R1 is set to 2, no SMARTS are produced at
all because the structure contains only one R1 node (i.e. the MDL file
does not make sense as a query).
If the RestH condition is ON (and the occurrence count at the default
>0) the SMARTS
[C;!h0;!h1;!h2][N;D2]c1ccccc1
is produced, which matches only the first structure. In order to retrieve
the second structure also, the query has to be changed to
with the RestH condition ON, and the Occurrence Count at the default >
0. This results in the two SMARTS
-
[C;!h0;!h1;!h2][N;D3](c1ccccc1)c1ccccc1
-
[C;!h0;!h1;!h2][N;D2]c1ccccc1
which respectively match the second and first database structures. The
D attributes on the N atom prevent matches against the third database structure;
such a match is excluded by the R-Group logic in the MDL file which requires
each R-group to be either Ph or H (and nothing else).
5.1.4 Zero Occurrences
In MDL RGfiles it is possible to build queries with extremely complex exclusion
conditions for R-groups, dependent on the values taken by other R-groups.
For example:
R1--C==C--R2R3
R1 = Cl
R2 = NO2
R3 = NO2 or SO3 or COO
R1 >0
if R1 then R2
R2 = 0
R3>0
This means that if the first C is substituted by Cl (R1) then the condition
for R2 must be satisfied. Because occurrence of R2= 0 this means that in
this case there must be no nitro group on the second C. If there is no
chlorine at R1, then R2 may be anything and the second C can be substituted
by any alternative of R3. Three SMARTS are produced for this query:
-
[C;D2] (=[C;D2] [Cl]) [!$(N(C=C)(=O)=O);
$(N(C=C)(=O)=O), $([#8](C=C)S(=O)=O), $(C(C=C)([#8])=O)]
-
[C;D2] (=[C;D2] ![Cl])
[$(N(C=C)(=O)=O), $([#8](C=C)S(=O)=O), $(C(C=C)([#8])=O)]
-
[C;D2] (=[C;D1])
[$(N(C=C)(=O)=O), $([#8](C=C)S(=O)=O), $(C(C=C)([#8])=O)]
SMARTS (1) deals with the situation where R1 is Cl, and excludes the possibility
of R2 being NO2 in AND (;) relationship with the alternatives for R3 (thus
effectively making the first alternative for R3 irrelevant since it conflicts
with the exclusion, but the SMARTS as a whole remains valid). SMARTS (2)
deals with the situation where R1 is not Cl, in which case all the values
for R3 are possible. SMARTS (3) deals with the possibility that R1 is H
(specified by the D1 attribute on the second carbon), and allows all three
possible values for R3.
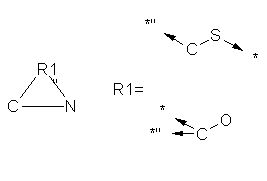
5.1.5 R-groups with variable positions
R-groups with variable positions are shown in RGfiles by placing an R-group
at each possible position, and using the Occurrence Count specification
to restrict the number of them. SMARTS does not have an equivalent feature,
and separate SMARTS patterns are generated for each possible combination
of R-groups. In some cases this can result in a very large number of SMARTS
being produced. For example in the following structure:
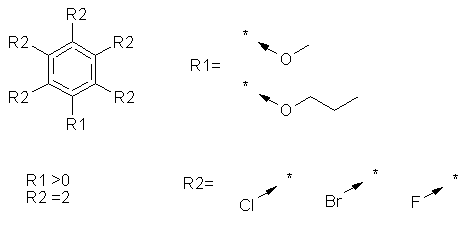
80 separate SMARTS are produced (there are 10 (= 5 x 2) ways of putting
two R2 groups at the five available positions and 8 (2 x 2 x 2) ways in
which the three unfilled positions can each be either a negative specification
or a hydrogen). The first two SMARTS produced are:
[c;D3]1 ( c ( [c;D3] ( [c;D3] ( [c;D3] ( [c;D3]1- [ Cl,Br,[#9] ] )
- [ Cl,Br,[#9] ] ) - [![Cl];![Br];![#9]]) - [!Cl;!Br;![#9]]) -
[$([#8](-c(c)c)-[#6]), $([#8](-c(c)c)CC[#6]) ] ) -[!Cl;!Br;![#9]]
[c;D2]1 c ( [c;D3] ( [c;D3] ( [c;D3] ( [c;D3]1-[Cl,Br,[#9]] )
- [Cl,Br,[#9]]) - [!Cl;!Br;![#9]]) - [!Cl;!Br;![#9]] ) -
[$([#8](-c(c)c)-[#6]), $([#8](-c(c)c)CC[#6])]
MOLSMART is configured to generate a maximum of 200 separate
SMARTS strings; if the conversion requires more than this a warning message
is given, and just the first 200 are output. (See Section
7.5 Program Limitations)
5.2 R-groups with 2 Attachment points
As with "Link Atoms" (see Section 4) two different
procedures are used for conversion of Rgroups with two attachment points.
If the SMARTS which starts at the R-group and finishes at the end of the
branch is self-contained a recursive SMARTS is created; otherwise all alternatives
are enumerated as separate SMARTS strings.
The two attachment points on an R-group are orientated as follows: the
first attachment point of the R-group is the atom in the root whose bond
to the R-group occurs first in the Bond Block of the MDL Ctab. The second
attachment point is always realised by a "ring closure" bond.
6. Conversion of Reactions
If the input MDL file to be converted is an Rxnfile describing a reaction,
MOLSMART
normally generates a Reaction SMARTS pattern. If the "-k" or "-m"
argument has been specified (either on the command line for single file
conversion - see Section 2.1 - or following the filename
in interactive mode - see Section 2.2), it will generate
a SMIRKS string, provided the reaction conforms to the special requirements
for SMIRKS.
6.1. Reaction SMARTS
If the input file is an MDL Rxnfile and the -k option has not be used,
MOLSMART
produces a reaction SMARTS. Thus the following reaction

generates the Reaction SMARTS:
([#6]CC(=O)O[#1]).(O([#6])[#1])>>([#6]CC(=O)O[#6]).(O([#1])[#1])
If any reactant/product atom mappings are shown in the Rxnfile these will
be retained in the reaction SMARTS.
6.2.SMIRKS without automatic atom mapping
If the -k option has been specified MOLSMART will check that the
reaction shown in the input file conforms to SMIRKS requirements, and will
generate a SMIRKS string as output; any atom map classes which are specified
in the input file will be reproduced in the SMIRKS. MOLSMART
will give an error message if the input file does not conform to SMIRKS
requirements, but from MOLSMART version 2.1 only the reduced restrictions
on SMIRKS introduced at Daylight
version 4.61 are imposed. The SMIRKS generated will be
identical to the reaction SMARTS which would have been generated without
the -k option, the only effect of the -k option being to check for features
(such as variable bond types) which are still not permitted in SMIRKS.
Though unbalanced reactions and incomplete atom maps are permitted,
it should be noted that unmapped atoms in SMIRKS are assumed to disappear
(into unspecified by-products) from the reactant side and to appear (from
unspecified reagents) on the product side. Map classes for corresponding
atoms on the reactant and product side should therefore be included in
the input file. Also, because SMARTS expressions are not permitted
for unmapped atoms in SMIRKS, if the input file contains any List Atoms,
or generic atom types (A and Q), these must appear on both sides of the
reaction and be mapped. Alternatively, MOLSMART's
automatic atom-mapping mode can be used.
6.3 Automatic Atom Mapping
If the -m option has been specified MOLSMART will automatically
calculate atom maps for any atoms which do not have them in the input file
(atom maps specified in the input file will be retained, though the actual
numbers used may change). In the above example, the resulting SMIRKS string
is:
([#6:1][C:2][C:3](=[O:4])[O:5][#1:6]) . ([O:7]([#6:8])[#1:9])
>> ([#6:1][C:2][C:3](=[O:4])[O:5][#6:8]) . ([O:7]([#1:9])[#1:6])
MOLSMART is able to assign atom maps automatically only if the reaction
is fully balanced, or is missing only a trivial reagent or by-product (H2O,
EtOH, HF, HCl, HBr or HI). If it is not balanced, an error message
is displayed, and no SMIRKS is generated.
The algorithm used by MOLSMART to assign the atom maps is based on the
Principle of Minimal Chemical Distance
(MCD) which assumes that the result of a chemical reaction is achieved
by the redistribution of the minimum number of valence
electrons (minimum number of bond changes). If several different mechanisms
with identical chemical distances are possible,
then MOLSMART chooses one arbitrarily. If the user wants to specify
the mechanism this must be done by specifying map
classes for at least some of the atoms, or a "reaction centre status"
for appropriate bonds, and this information will be taken
account of by MOLSMART. Only atoms which carry the same atomic symbol
and weight can be mapped to each other.
Though MOLSMART is generally able to identify the mapping scheme corresponding
to the minimum chemical distance very
rapidly, in a very few cases it is unable to confirm rapidly that there
are no other mapping schemes with a lower chemical
distance. Rather than exhaustively trying all possible mappings , the
program generates a SMIRKS with the best mapping it has
so far found, along with a warning that this may not be optimal (i.e.
may involve more bond changes than are strictly necessary).
The mapping produced will still be valid, and will normally be optimal
as well, but confirmation of this can be obtained by
mapping at least some of the atoms manually.
6.4 Reaction Stereo Chemistry
In cases where the user has not specified any "atom stereo parity" but
has specified an "inversion/retention flag" two separate SMIRKS are produced.
Thus in the reaction:
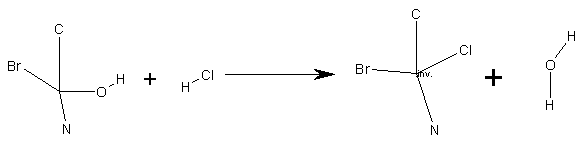
where the chirality on the central carbon is inverted, the following
two SMIRKS are produced, each representing one of the possible configurations
for the reactant and product:
-
([C@@:1]([#6:2])([#7:3])([Br:4])[O;h1:5]) . ([Cl:6][#1:7])
>> ([C@@:1]([#6:2])([Br:4])([#7:3])[Cl:6]) . ([O;h1:5][#1:7])
-
([C@:1]([#6:2])([#7:3])([Br:4])[O;h1:5]) . ([Cl:6][#1:7])
>> ([C@:1]([#6:2])([Br:4])([#7:3])[Cl:6]) . ([O;h1:5][#1:7])
7. Program Limitations
Most MDL queries can be translated precisely to one SMARTS string, though
when R-groups are present in the query or the "inversion/retention" flag
is set, more than one SMARTS string may be required (to be searched in
OR relationship). In a very few cases there may be some limitations on
the faithfulness of the translation.
The following features of MDL files are not handled at all:
-
custom P-tables,
-
3D queries
-
S-groups
The SMARTS strings generated by MOLSMART are not optimised for efficiency.
If being used for searching, it may be appropriate to reformat them using
the dt_smarts_opt function in the Daylight SMARTS Toolkit.
7.1 R-groups and Further Substitution
In MDL RGfile queries an R# node does not necessarily indicate just a single
node (which might consist of several atoms and alternatives) but it may
define the unfilled valences of the atom to which the R# is bonded. The
problem actually lies in the differing ways in which the search algorithms
used by MDL and Daylight interpret the information given in the query,
and thus in certain cases some compromises have to be made in the conversion.
MOLSMART automatically adds a "degree" descriptor to certain
atoms in the SMARTS (for examples see Section 5.1.1).
This forbids any further substitution at a node and is used where
-
a node is substituted by an R-group with RestH logic
-
a node is substituted by an R-group with IF R# THEN logic
-
a node is substituted with an R-group which appears more often in the query
than its minimal occurrence is.
In all cases where no further substitution is possible or wanted the logic
of the MDL query is precisely converted into SMARTS. However, if the user
wants to allow further substitution at these atoms, the query must be built
as several distinct MDL queries which can be separately converted to SMARTS
and ORed or ANDed together afterwards.
7.2 Multiple R-groups at a single site
In the RGfile data structure, up to 8 different R-group numbers can be
associated with a single R# node. MOLSMART allows only 2 different
R-groups to be associated with one R# node, and an error message is given
if more than two are associated with it.
7.3 Other Limitations of R-group Handling
MOLSMART is unable to handle combinations of Occurrence Count values,
such as
"4, 6-7"
MOLSMART cannot handle R-groups with two attachment points which
occur more than once
7.5 Size Limitations
As noted in Section 5.1.5 there is a limit of 200
separate SMARTS patterns which can be generated from a single RGfile input.
MOLSMART is also unable to handle MDL files containing more than
1000 lines, or with lines of more than 80 characters (the latter are not
permitted in MDL files in any case).
8. Support and Further Information