
Fast Calculation of Molecular Polar Surface Area Directly from SMILES
Peter Ertl, Bernhard Rohde, Paul Selzer
Novartis Pharma AG, ChemInformatics, CH-4002, Basel, Switzerland
peter.ertl@pharma.novartis.com
Introduction
 Molecular Polar Surface Area (PSA) defined as the sum of surface contributions of polar atoms (usually oxygens, nitrogens and attached hydrogens) in a molecule, has been shown to correlate well with drug transport properties, such as intestinal absorption, or blood-brain barrier penetration [1-7].
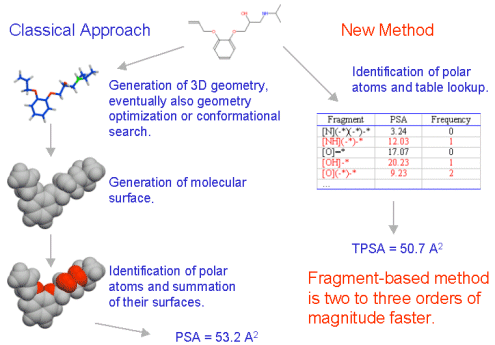
Current methodologies to calculate PSA, however, are relatively time consuming, because of the necessity to create a reasonable 3D molecular geometry and to calculate the surface itself.
In order to enable virtual bioavailability screening of very large collections of molecules or whole virtual combinatorial libraries, we developed a new, very fast methodology to calculate the PSA from fragment contributions. We termed this method TPSA - topological PSA.
Molecular Polar Surface Area (PSA) defined as the sum of surface contributions of polar atoms (usually oxygens, nitrogens and attached hydrogens) in a molecule, has been shown to correlate well with drug transport properties, such as intestinal absorption, or blood-brain barrier penetration [1-7].
Current methodologies to calculate PSA, however, are relatively time consuming, because of the necessity to create a reasonable 3D molecular geometry and to calculate the surface itself.
In order to enable virtual bioavailability screening of very large collections of molecules or whole virtual combinatorial libraries, we developed a new, very fast methodology to calculate the PSA from fragment contributions. We termed this method TPSA - topological PSA.
Methodology
The methodology for the calculation of the PSA from fragment contributions is described in detail in [8]. The procedure is based on the summation of tabulated surface contributions of polar fragments (i.e. atoms regarding also their environment). The fragment contributions were determined by a least squares fit to the single conformer 3D PSA for 34810 drug-like molecules.
In this way the surface contributions of 43 polar fragments centered on oxygen, nitrogen phosphorus and sulfur were determined. The full list of fragments together with their PSA contributions is published in [8]. The statistical analysis showed a very good correlation between 3D PSA and TPSA:
r = 0.991, r² = 0.982, sigma = 7.83 Ų, average absolute error = 5.62 Ų.
The workflow of the new methodology compared to the standard procedure to calculate PSA is shown on the scheme below.
Application of TPSA to the Prediction of Drug Transport Properties
The new TPSA methodology has been validated by correlation with published data of various types of drug transport properties, including intestinal absorption, blood-brain barrier penetration, and Caco-2 cell permeability. All these datasets have been already studied by using 3D PSA. In all cases, the TPSA methodology is performing well, providing results of practically the same quality as the computationally much more demanding 3D PSA. Details about validation studies are summarised in [8].
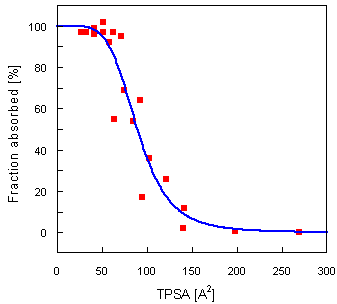
Here, as an example, a correlation between TPSA and intestinal absorption is reported.
A set of 20 representative drugs together with their absorption ratio after oral administration in humans have been collected by Palm et al. [1]. The authors found a strong sigmoidal relationship (r² = 0.94) between absorption and so called dynamic PSA, based on the Boltzmann-weighted average values computed from an ensemble of low-energy conformations obtained by a detailed conformational search.
Correlation with TPSA provides practically identical sigmoidal relationship with r² = 0.91 (see the graph below). The time requirements needed for the calculation of TPSA, however, are two to three orders of magnitude lower.
Move the mouse over the red points to see the corresponding structures.
|
|
Interactive Calculation of TPSA with Java Applet
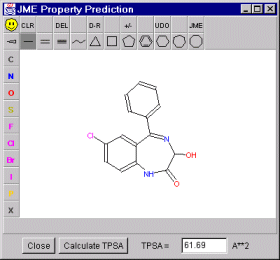 To enable fast interactive calculation of TPSA we developed a program with these capabilities.
Program enables easy input and editing of molecules and calculation of the topological PSA. The system is written in Java and may be used as an applet incorporated directly in the web page, or as a standalone application.
The program is written as an add-in module to the Novartis JME Molecular Editor applet.
To enable fast interactive calculation of TPSA we developed a program with these capabilities.
Program enables easy input and editing of molecules and calculation of the topological PSA. The system is written in Java and may be used as an applet incorporated directly in the web page, or as a standalone application.
The program is written as an add-in module to the Novartis JME Molecular Editor applet.
You can experiment with the interactive calculation of the TPSA by clicking the image on the right.
Daylight Toolkit Contrib Program for TPSA Calculation
The Java applet allows calculation of TPSA, but this interactive procedure is only practical for data sets of standard size (up to several dozens of molecules). For the calculation of the TPSA for large collection of molecules we developed a C program based on the Daylight Toolkit. The program reads list of molecules encoded as SMILES and calculates the topological PSA for them.
The TPSA calculated by this program and by the JME applet may differ slightly for some classes of molecules. This is caused by the different interpretation of aromaticity in the JME system and in the Daylight software.
Conclusions
A new methodology for the calculation of the molecular polar surface area as a sum of fragment contributions was developed.
The method is straightforward and does not require any computationally demanding steps such as 3D structure generation or surface calculation.
The only input needed is the molecular topology (i.e. SMILES string).
With a throughput of about 20 000 molecules per minute the computational speed of the new approach is two to three orders of magnitude faster than currently used methods, so it can be applied to virtual bioavailability screening of very large collections of molecules.
Despite the approximations, the results are of comparable quality to those obtained by using computationally much more demanding approaches, as documented by the validation studies with published drug transport data, including intestinal absorption and blood-brain barrier penetration.
References
- Palm, K., Stenberg, P., Luthman, K., Artursson, P. Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm. Res. 1997, 14, 568-571.
- Palm, K., Luthman, K., Ungell, A.-L., Strandlund, G., Artursson P. Correlation of drug absorption with molecular surface properties. J. Pharm. Sci. 1996, 85, 32-39.
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807-814.
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J. Pharm. Sci. 1999, 88, 815-821.
- van de Waterbeemd, H., Camenisch, G., Folkers, G., Raevsky, O.A. Estimation of Caco-2 cell permeability using calculated molecular descriptors. Quant. Struct.-Act. Relat. 1996, 15, 480-490.
- Winiwarter, S., Bonham, N.M., Ax. F., Hallberg, A., Lennernäs, H., Karlén, A. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J. Med. Chem. 1998, 41, 4939-4949.
- Kelder, J., Grootenhuis, P.D.J., Bayada, D.M., Delbressine, L.P.C., Ploemen, J.-P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514-1519.
- Ertl, P., Rohde, B., Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714-3717.