5. SMIRKS - A Reaction Transform Language
A transform is simply a generic reaction within the Daylight system. Generic reactions are extremely useful for chemical information processing. They can be used to create new reactions, manipulate molecules, and to generate new molecules on a large (conbinatorial) scale. They are somewhat complicated, because they must meet several conflicting sets of requirements. These requirements, and how the Daylight system addresses them, are discussed here.
5.1 Description
A complete reaction can be described in a number of ways: the reactant/product notation used in SMILES, as a reaction graph, and as a atom- and bond-change list plus the reactant molecules. It is straightforward to interconvert any of the three representations of a complete reaction.
The most intriguing way to think about a complete reaction is as an atom and bond change list. This representation can be easily understood to capture the idea of a generic reaction. Any set of reactions which undergo the same set of atom and bond changes, regardless of the underlying molecule substrates, can be considered an example of a given generic reaction.
Consider once again our Sn2 reaction. If it is a normal Sn2 reaction, then the list of atom and bond changes during the reaction is as follows:
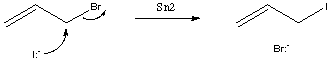
| Reaction Change List: | ||
| Part: | Change Type: | Change: |
| C-Br | Bond | Single bond -> no bond |
| C-I | Bond | no bond -> Single bond |
| Br | Atom | no charge -> -1 charge |
| I | Atom | -1 charge -> no charge |
Any reaction which undergoes the same set of atom and bond changes would be considered part of the same generic reaction. For example, reaction of Potassium Iodide rather than Sodium Iodide, or reaction of any alkyl bromide in place of Allyl Bromide all have the same list of atom and bond changes. Note that this bare-bones representation of the reaction does not take into account other factors which might affect the reaction such as the steric effects of a primary bromide versus a secondary or tertiary one, and the electronic activating effect of the allylic bond.
As an aside, note the similarities of this representation with the "Difference Fingerprints" described previously. In effect, the difference fingerprint is calculated directly from the bond changes during a reaction (atom property changes like charge, stereochemistry, are not included). The difference fingerprints will be identical for all examples of a single generic reaction.
So, there are two distinct requirements to accurately capture a generic reaction. First is the actual set of changes to the molecule which occur during the reaction (captured with the atom and bond changes) and second is the indirect effects of activating and deactivating groups near the reaction site.
Within the Daylight system, the indirect effects on a generic reaction are most appropriately expressed with the SMARTS query language. With it, one can express concepts such as "electron-withdrawing group", "electron-donating group", aromaticity, unsaturation, steric effects, etc.
The parallels here should be evident: a complete reaction consists of a set of atom and bond changes, plus the substrate molecule upon which the changes operate. A generic reaction consists of the same set of atom and bond changes, plus a substrate SMARTS pattern upon which the changes operate. Any molecule which matches the SMARTS pattern is a candidate for the generic reaction.
5.2 Representation
In the Daylight System, we adopted the reactant/product notation for generic reactions. It is not as compact as a reaction graph, but it is the most compatible and most consistent with the SMILES and SMARTS languages already defined for reactions.
The language SMIRKS is defined for generic reactions. It is a hybrid of SMILES and SMARTS in order to meet the dual needs for a generic reaction: expression of a reaction graph and expression of indirect effects. It is a restricted version of reaction SMARTS involving changes in atom-bond patterns. The rules for SMIRKS are:
- The reactant and product sides of the transformation are required to have the same numbers and types of mapped atoms and the atom maps must be pairwise. However, non-mapped atoms may be added or deleted during a transformation.1
-
Stoichiometry is defined to be 1-1 for all atoms in the reactant and product for a transformation. Hence, if non-unit stoichiometry is desired, reactants or products must be repeated.
-
Explicit hydrogens that are used on one side of a transformation must appear explicitly on the other side of the transformation and must be mapped.
-
Bond expressions must be valid SMILES (no bond queries allowed).
-
Atomic expressions may be any valid atomic SMARTS expression for nodes where the bonding (connectivity & bond order) doesn't change.1 Otherwise, the atomic expressions must be valid SMILES.
1 Introduced in v4.6.1.
These above rules guarantee that the SMIRKS can be interpreted as a reaction graph and that the atom and bond changes can be derived from this representation. This set of rules satisfies the first requirement for a generic reaction. The final rule allows the expression of the "indirect effects" of a generic reaction for atoms which don't participate directly in the reaction.
The net result of these rules is a language which can capture the ideas of generic reactions. A number of examples follow to illustrate features of the language.
5.3 Transform Grammar
transform : reactant '>' agent '>' product | reactant '>>' product ; reactant, agent, product : pattern ; pattern : SSMARTS ; SSMARTS: a valid pattern specification, excluding bond expressions, and using a limited set of atom expressions (Subset-SMARTS).
5.4 Examples
First is a simple transform to interconvert nitro-group representations in the toolkit. The nitro group is typically represented either with pentavalent Nitrogen "*N(=O)=O" or as the charge-separated trivalent Nitrogen "*[N+](=O)[O-]". These can be interconverted with the following transform:
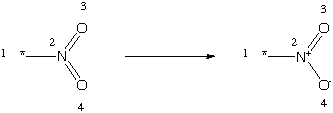
[*:1][N:2](=[O:3])=[O:4]>>[*:1][N+:2](=[O:3])[O-:4]
This transform illustrates an important point: transforms need not represent real reactions. Transforms are useful as a general tool for manipulation of molecules in the toolkit. Most atom and bond changes can be written as legal transformations. Hence, transforms become a powerful tool in the chemist/programmers arsenal for chemical information processing. Also, as with any transform, this one can be used in either the forward or reverse direction.
Inspection of the transform indicates that this meets the requirements for a legal transform. First, it has the same number of atom expressions on both sides of the transform, and they are mapped pairwise. The atom expressions are all legal SMILES and the bond expressions are all legal SMILES.
In this example, there are no SMARTS expressions found. SMARTS atomic expressions could be substituted for the atoms of map classes ":1" and ":3" only. The two atoms attached to the bond which changes ("N:2" and "O:3") must be expressed as SMILES. The change in valence and charge which occurs can be deduced unambiguously from the SMIRKS. Were atomic expressions allowed for these nodes, the determination of atomic properties might not be possible.
The next example illustrates the most confusing part of the SMIRKS language, which is the handling of hydrogens. Unfortunately, the SMILES and SMARTS languages express hydrogens inconsistently. These inconsistencies have been partially reconciled in the SMIRKS language by first, requiring that all hydrogens directly involved in a transform (bonds change) must be expressed explicitly and second, changing the meaning of SMARTS for a single case: [H]. There are still some cases which will cause confusion, however.
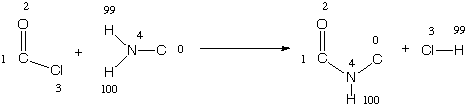
[C:1](=[O:2])[Cl:3].[H:99][N:4]([H:100])[C:0]>> [C:1](=[O:2])[N:4]([H:100])[C:0].[Cl:3][H:99]
Note that both hydrogens attached to the nitrogen of the reaction are shown as explicit. Based on the SMIRKS rules, the expression [H:99] must be interpreted as SMILES, since the bonding to this node changes during the reaction. The expression [H:100] may be interpreted as SMARTS, since its bonding does not change in the reaction. Recall that in versions of the Daylight system prior to 4.51, [H] as SMARTS meant: "any atom with a single attached hydrogen", while in SMILES it is a lone explicit hydrogen.
These differences in interpretation would make SMIRKS unintelligible. Hence, a single change to SMARTS interpretation, for expressions of the form: [<weight>]H<charge><map>]. In SMARTS, these expressions now are interpreted as a hydrogen atom, rather than as any atom with one hydrogen attached. All other SMARTS hydrogen expressions retain their pre-4.51 meanings.
| SMARTS/SMIRKS hydrogen expressions: | ||
| Expression: | 4.42 meaning: | 4.51 meaning: |
| [H] | Atom with one attached hydrogen | A hydrogen atom |
| [#1] | A hydrogen atom | Unchanged |
| [H1] | Atom with one attached hydrogen | Unchanged |
| [*H] | Atom with one attached hydrogen | Unchanged |
| [H,+] or [*,H], etc. | Unchanged | |
The result of the change in semantics is that both explicit hydrogens in the example SMIRKS are interpreted consistently as hydrogen atoms. Note that there still may be confusion for 'implicit' hydrogens. For example, if the amide formation reaction were expressed as:
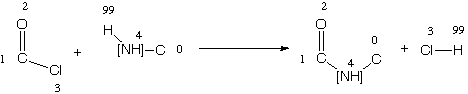
[C:1](=[O:2])[Cl:3].[H:99][NH:4][C:0]>> [C:1](=[O:2])[NH:4][C:0].[Cl:3][H:99]
This case only matches secondary amines. The expression [NH:4] matches a nitrogen with exactly one hydrogen attached (the [H:99] is it). Hence, any other attachments must be non-hydrogen. In general for SMIRKS, the best strategy for expressing hydrogens is to include them as explicit atoms if they are involved in the reaction directly or if they are attached to atoms which are involved in the reaction. This will eliminate most of the confusing cases.
Stereochemistry in SMIRKS is handled locally based on atom map labels. That is, a stereochemical specification describes the orientation of atoms or bonds based solely on the ordering in the string and the atom map labels. For example:
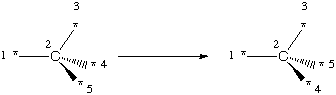
[*:1][C@:2]([*:3])([*:4])[*:5]>>[*:1][C@@:2]([*:3])([*:4])[*:5]
[*:1][C@:2]([*:3])([*:4])[*:5]>>[*:1][C@:2]([*:4])([*:3])[*:5]
This inverts any carbon stereocenter encountered. On the reactant side of the transform, the expression describes a specific orientation of the nodes; similarly, on the product side the inverted orientation of the same nodes is described. Similarly, for bond stereochemistry:
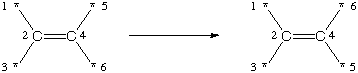
[*:1]/[C:2]([*:3])=[C:4]([*:5])/[*:6]>>[*:1]/[C:2]([*:3])=[C:4]([*:5])\[*:6]
This inverts any C=C double-bond stereochemistry matched. Note that both of the above examples can match a single stereocenter multiple ways, however the net result is always an inversion of the stereocenter based on the specification of the transform.
In general, transforms which involve stereochemistry should be written with sufficient context for the toolkit to interpret the local chirality needed for analysis. For tetrahedral chirality, all four connections to the chiral atom should be explicitly shown and for double-bond chirality, all three connections to each atom (one double-bond and two single bonds) should be shown.
Finally, a point about the new component-level grouping operators in SMARTS and SMIRKS. This syntax allows the expression of inter- and intramolecular reactions in both SMARTS and SMIRKS. This syntax is fully supported in SMIRKS. See the section on SMARTS section on Reaction Queries for more information.
Go To Next Chapter... 6. Fingerprints - Screening and Similarity