
It's my opinion that any discussion or study of History is dull and uninteresting--unless it gives insight into motives and objectives and how these may have changed over time. These mottos are somewhat 'corny', but they do express the idea that at the Pomona College Medchem Project, where this all began, we did not want to build a Black Box calculator. We wanted CLOGP to make it clear to the user what its calculations were based on. And when Dave Weininger came on board the Medchem Project, we found him to be a 'kindred spirit' in this regard. In fact, he could sometimes be annoying when he would say: "Al, you told me that you wanted this Interaction Factor to apply to 'such and such' a structure, but, stated that way, it would also apply to these other structures. Do you have any measurements to support that?" And then we would have to get some of that solute to measure, or else restrict the original statement of the rule. But we tried our best to keep CLOGP based on known solvation forces, especially those expressed by the Solvatochromic parameters being developed at that time by Kamlet and Taft in the U.S. and Taylor, Abraham and Leahy in England. As I will point out later, most of the log P programs which have come on the scene lately rely on statistical methods in their construction as well as their validation. Surely old Mark Twain had 'tongue in cheek' when he disparaged statisticians, but we shouldn't lose sight of the solvation forces when we build a calculating method that is supposed to predict the partitioning equilibrium. We may need statistics to keep us in line, but we should be, first and foremost, physical chemists.

Four years before I came to work with Corwin Hansch at Pomona College, he and Fujita developed the first method of calculating log P from structure based on adding a substituent =BC value to the measured log P of a parent. When additivity failed, using =BC values from benzene, they saw tha= t the =BC scale was dependent on the kind or ring involved; i.e. whether the substituent was being placed on an aromatic ring was electron rich or electron poor. This data enabled them to propose reasonable mechanisms for the differential solvation in the two phases. But the method depended upon having a measured value for every 'parent' structure one wanted to work with. So I began working on a method which 'started from scratch.'
Before we were ready to disclose this work, Ralph Rekker and his colleagues published a "Fragmental" method of calculating log P (octanol). His was a manual method which depended a great deal on the user's prior knowledge to choose the proper multiplier for the "Magic Constant" which governed polar interactions. Yet it became widely used, and is still referenced frequently. Dave and I wanted to program a 'fragment' method which would take apart any structure without user input, and then look in updatable tables to see if the necessary data was there to re-assemble the fragments, making sure to account for any interaction there might be between them. We had drug and pesticide developers in mind primarily, and thus we proceded rather cautiously, hoping that a "missing fragment" message would induce the company which was investing a sizable sum to develop a lead, to measure some key partition coefficients so they could encorporate the new data in their versions of the program through Algorithm Manager. This hope was actuallized in some instances, and, in rarer instances, the new data was made available to us to distribute within CLOGP.
At this point we received some welcome financial support from the U.S. EPA who learned (largely through the work of Brock Neely and his colleagues at Dow Chemical) that log P was a good predictor of bioaccumulation and useful in environmental models of solute transport. But for their needs, it was not necessary to know why a chemical had the log P it did. The chemical structures were a given; they just needed to know what was the log P of each. Perhaps co-incidentally, there appeared in the marketplace quite a number of computerized log P calculators. While it was gratifying to note that many (if not most) made use of subsets of the Medchem Starlist which we had widely distributed, it was somewhat disconcerting to see that in the construction these programs more attention was paid to statistics than to physical chemistry.

Statistics certainly has its place in testing what you've done, but when 'push comes to shove' in deciding which path to follow in development, we want it to make sense physically, even if the statistics suffer somewhat. We were fortunate to work closely with the group begun by Kamlet and Taft who were developing Solvatochromic parameters which highlight the importance of solute size polarity and hydrogen bond acceptor strength.

This work was continued by Taylor, Abraham and a group at Nantes, France who examined partition coefficients from other solvent systems which respond differently from octanol in accomodating solute polarity. These studies also confirmed the Hansch-Fujita postulate that octanol and water were equal in HBD strength. This collaboration also helped us deal with the non-additivity of these effects. These four diagrams illustrate the contribution of each of the four parameters as they act upon the solute, 4-amino-pyridine.

N-methyl-Isoniazide illustrates non-additivity in the important HBA strength parameter. HBA of both the pyridine nitrogen and the terminal NH-methyl are reduced by the carbonyl's electron withdrawing power. And the Methylhydrazine function partly shields access to the oxygen's two lone pairs. It is factors like these that make fragment estimation from parts smaller than used in CLOGP a dangerous exercise.

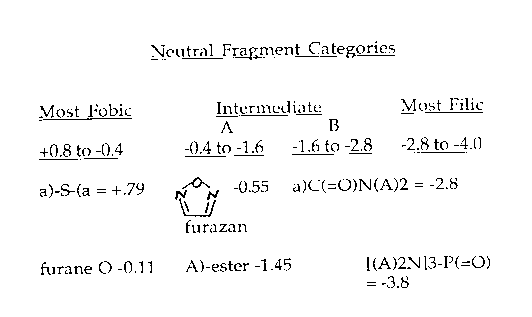
Comparing the performance of log P programs is no easy task, as I believe Yvonne Maritin will attest in her presentation. Most reports I have seen in the literature suffer from using too small a test set, especially when it also contains erroneous values. One such fairly recent report by Mannhold & Dross (QSAR 15, 403 '96) examined 13 calculation methods for 138 solutes. This represents a monumental effort,(only part of which is shown here) but at least four of the drug molecules were measured at a pH 4.5 log units on the ionic side of the pKa. In 'correcting' for ionization, the assumption was made that only the neutral form was present in octanol, when actually that phase contained more ion pair than neutral solute. Just four very bad values in a set of this size can distort the statistics enough to cast some doubt on the conclusions reached. Unfortunately errors like this take on a life of their own. Moriguchi used a subset of 22 of these drug structures, to validate his methodology, and his conclusions were affected to an even greater extent. I published a note in J. Pharm.Bull.,pointing out this error, but I fear it will have little effect. I recently got another paper to review which quoted the same 138 datapoints supposedly validating yet another new methodology.


At present it looks like the Standard Set will contain very nearly 10,000 structures, about two-thirds of which we consider to be relatively simple and should be calculated with an average deviation of 0.2 or less. The other one-third are complex with multiple interactions between polar groups, some at quite a long topological distance. It will take a rather clever program to calculate the entire set with an average deviation less than 0.3 log units.
It would be difficult to devise a fragment system of log P calculation that gives more accurate estimations than the original substituent-=BC method of Hansch and Fujita. But as I mentioned earlier, there were not enough measured 'parent' log Ps to make the =BC method feasible for calculating the structural files of 30 years ago. The size of structural files has taken a quantum leap since then, due both to combinatorial chemistry, and natural product chemistry, and we now find that our conservative approach of manually estimating CLOGP's 'Missing =46ragments' is not keeping pace. It is inhibiting the Clustering methods which are needed to make sense of this new 'meteoric shower' of facts. Up to this time, in estimating new fragment values, we first look for the closest measured fragments and what effect the needed changes on these might have on the Solvatochromic parameters of polarity and hydrogen-bond acceptor strength. We have added quite a few of these already, giving them an error level of 40. Obviously this approach time-consuming, and we cannot anticipate all the fragment structures that CLOGP might encounter. =46ortunately, for clustering purposes, as oppposed to optimizing drug design, the required accuracy of log P prediction is less. Later I will describe a Fragment Estimation method under development to be used when reliability requirements are less stringent, and these values will depend largely on statistical validation. Even if our values for these 'Unmeasured Fragments' are no more accurate than those other methods claiming 'No Missing Fragments', CLOGP will have one great advantage: We can make use of the present framework for estimating electronic interactions (i.e. sigma/rho and always positive) and also ortho interactions which can be either positive (H-bonding) or negative (steric 'twisting).
Obviously a program does not 'earn its keep' just because it can calculate structures which are already available in a measured database or are simple homologs of measured structures. It must 'extrapolate' quite a ways into 'unexplored' hydrophobic space. There is NO substitute for measurement, but when measurement is impractical we must ask: "What price am I willing to pay to get 'No Missing Fragments'?"


I cannot keep up with the performance of 13 programs like Mannhold and Dross attempted to do, so I just look at two which have gained some popularity and which operate on different principles from CLOGP and from each other. They are the ACDlogP program from Advance Chemistry Development in Toronto, and the KOWWIN program from Syracuse Research. Both utilize a database of measured fragments, and both attain more flexibility than CLOGP by allowing the 'fusing' of fragments together, usually accompanied some correction factors designated by special atom-strings. From my experience I would guess that 60% of the time such 'Unmeasured Fragments' are accurate to =B10.6, which seems adequate for clustering purposes. Now I will point out some risks that must be faced with the other 40%.
Hexazinone was present in our Medchem database before we had a measured log P for it. Following our cautious policy, CLOGP returned the 'missing fragment' message until we found a reliable measured value of +1.85. We then entered the value for the triazinedione fragment after allowing for an electronic interaction with the tert.amine substituent. Obviously the CLOGP calculation is good for this structure, but note that it allows for a different electronic interaction if the dimethylamino group is replaced by, say, a nitro, and also it will automatically compensate for any polar function attached to the cyclohexane ring.
ACDlogP, following its own rules for 'fragmentation', includes the amino nitrogen in a large fragment which contains all the ring hetero atoms. After doing this, it then 'sees' a guanidine substructure with an almost unrealistically low value of -6.39. Then it warns that measurement must be made at a very high pH to get a 'neutral' log P. (Of course the nearby carbonyls cancel the solutes's basicity.) Based on its past experience, KOWWIN makes several overlapping correction factors which over-corrects for interaction. Of course when the measured value is discovered, it will be possible to give higher priority to other atom-strings which will bring the calculation in line.
If we had attempted to calculate the triazinedione fragment 'manually', we would have noted that, with five HBA sites, it had the potential of being quite negative: 1) In a totally aliphatic environment, one would guess it to be in the most negative category (as we will see later) i.e about -3.8. 2) However, by Huckel's Rule, the ring is aromatic and this would raise any fused-in fragments by .7 or .8. So the estimated value is closer to -3.0 The measured value turns out to be -2.79.
To enable CLOGP to make estimations of this type without outside assistance, we plan to set up four ranges of fragment values: A 'Most =46obic' a 'Most Filic' and two intermediates. Careful examination will be made of the current populations in each of these classes with respect to the number and type of HBAs in each fragment; the number and type of internal and external bonds; and overall fragment size. When CLOGP dissects a structure according to its fixed rules and encounters a polar fragment not in the Standard Fragment Database, it will apply the 'fragment estimation' rules to place it in one of these four ranges. As experience is gained, we believe that it will have sufficient 'insight' to place some of the newly encountered fragments, not just at mid-range, but at low or high portion of the 1.2 log unit range. This procedure should enable CLOGP to estimate new fragment values at least as accurately as the other methods we have seen, but also, by noting the external bonds of each new fragment, it is prepared to estimate electronic interactions with other polar fragments as I pointed out previously.
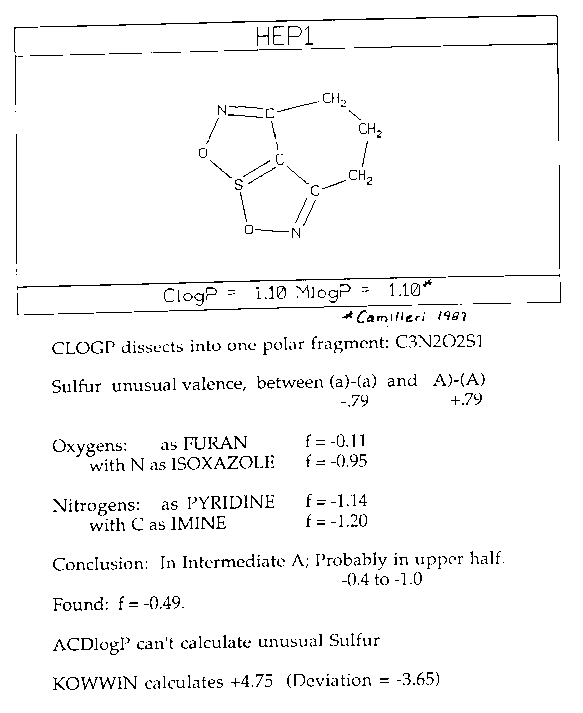
I have an early version of ACDlogP, and it reports that it cannot deal with a four-valent sulfur, and so it fails in this calculation. (It does deal with sulfoxides!) The Syracuse Research KOWWIN program sees the three carbons as extra hydrophobic, resulting in a serious overshoot.
SUPPLEMENTAL

CLOGP defines the 'new' fragment as circled in dotted blue lines. The sulfur and sulfonamides already appear in the measured Frag.DB. In estimating fragment hydrophobicity, the computer 'sees' an isothiazole substructure at -0.58; a nitrogen at the fusion of two rings at -0.56; an aromatic attached imine at -0.86; and a pyridine-type nitrogen at -1.14. The average of these is -0.78. But the fragment has an external bond to a phenyl ring, and thus for these analogs the estimated fragment value should be raised by 0.9 to +0.12.
The electronic nature of this fragment must now be estimated. Electron releasing or withdrawing substituents, X, will affect log P differently depending on the sigma/rho assignment to the new fragment. Position 8 will be most similar to the 2 or 5 positions of thiadiazole or the 2- position of pyridine. These are in close agreement, and usually it is best to take the lesser. The 2- position should be similar to an imine. CLOGP is now aware of possible electronic interactions and calculates these nine analogs very well, all things considered. It does give strong indication that measurement of the 4-NMe2 analog should be repeated.
ComFA and similar programs have created a need supposed for 'Hydophobic Field Analysis'--that is, establishing the hydrophobic nature at each portion of any solute structure. I worked with Don Abraham and Glen Kellog in the early development of their HINT program, but I have not attempted to verify its effectiveness. The CLIP program from Lausanne also claims to do this effectively. However, there are good theoretical reasons to believe that, at best, these attempts have serious limitations. And the 'Atom-Based' log P calculations, which also claim utility in this application, are totally misleading, at least in my opinion.
Broto initiated this 'atomic' approach in 1984 and Ghose, Crippen, and Viswanadhan have pursued it since. Their methods examine each atom in a structure on the basis of its oxidation and hybridization state, and then extends this to each of its neighboring atoms. Carbon atom types are identified with the familiar designations: sp3C; sp2C etc. The casual reader might assume that some fixed values from quantum chemistry or molecular orbital theory accompanies the atoms so designated. Not so. Hetero-atoms of all types are lumped together as X, whether they are small and good HBAs, like oxygen, or large and poor HBAs like sulfur and selenium. They are merely labels for variables that can be manipulated via multiple regression analysis. I have published ojections to their 1989 version, giving as an example the fact that their method sees the carbons in an aliphatic ether as hydrophilic and the oxygen as hydrophobic. In revising their 'atomic values' there must be little if any constraint supplied by theoretical chemistry, as the example, aniline, shows:
In the 1989 version the nitrogen atom was seen as quite hydrophobic, while the hydrogens (presumably acting as H-donors) gave the amino group its polar nature. The carbons in the phenyl ring were hydrophobically neutral, the ring hydrophobicity arising from their attached hydrogens. To bring their system more in line with Solvatochromic findings, their revised atom values now declare that the hydrophilic character of the amino group does, indeed, depend mostly on the nitrogen. But somehow the phenyl carbons have become strongly hydrophilic while their attached hydrogens are now very hydrophobic. A critic might say that we are seeing statisticians at play, not chemists looking for understanding.
Triflubenzuron, a chitin synthetase inhibitor having insecticidal properties, shows how CLOGP might be upgraded to more easily provide useful insights into localized hydrophobic/hydrophilic fields. CLOGP happens to give an extremely good estimate of overall hydrophobicity. But there may be much more valuable information hiding in the calculation details. Presently it takes the user valuable time to determine which is Ring #1 and which Ring#2, and even more time to ascertain the amounts and location of the ortho corrections. We ought to supply the means to dislplay these in the Depiction window just as I have in this overhead.
To make my point, I would like to speculate that the target site in the enzyme has been shown to have a large hydrophobic area opposite to a polar region which has one or more strong hydrogen bond donors. Adding hydrophobic groups to the meta and para positions of the two rings might add to hydrophobic binding in that area. On the other hand, fluorines in the ortho position have a low =BC value and furthermore they decouple the carbonylurea function from the ring, raising the HBA of the two carbonyl oxygens. Thus the -1.27 total ortho effect is really seen on these oxygens. This information could conceivably be of value in the design of better insecticides.