
A precursor set is chosen to maximise the diversity of coverage of 3D pharmacophore space. This space is defined in terms of the 3D geometric distribution of points of potential hydrogen bonding, charge interaction and hydrophobicity. A description of a monomer in these terms represents the contribution that monomer could make to binding to a receptor. Since the sets are intended to be re-useable in many libraries, we cannot measure the diversity of any resulting library. Instead, this measure seeks to maximize the pharmacophore space explored in a fixed position and direction from a core, or from a second monomer in a polymeric type library.
For these two structures, the potential pharmacophore points are labelled. Note that the substructure shown in blue has been added to the two primary alcohols to provide a fixed "core"
In 3D, considering only the pharmacophore points and their geometric relationships, the two structures appear as shown below.
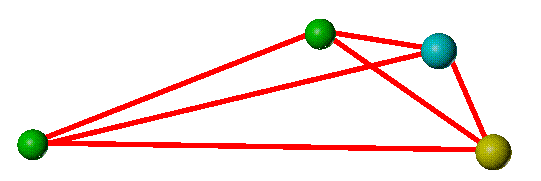
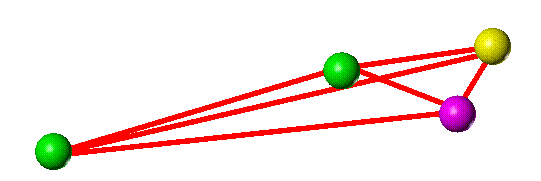
The two structures present different patterns of pharmacophore points relative to the "core" (represented by the two aligned green point-of-attachment markers) and so are considered to span different areas of space.

The scope of a precursor set is defined by the type(s) of chemistry to be done on the precursor and by what is commercially available. Compounds with functional groups which are more reactive than the target group must be excluded. Groups which would make any product compounds uninteresting as leads for further medicinal chemistry are also excluded. Two more straight forward scope definitions are as follows:
1) All commercially available primary alcohols with the exception of those structures with:
2) All commercially available carboxylic acids with the exception of those structures with
These scope definitions are translated directly into SMARTS definitions for searching the Available Chemicals Directory using MERLIN.
Whether the precursor set is chosen for a specific library or is to be reused many times it is necessary to make changes so that it has the structure that it will have in any products. In particular it must have the correct
If the set is re-useable, a "pseudocore" is added. This must be
For example:
1) Primary alcohols

2) Hydrazines

Once attached, the pseudocore provides a fixed reference point from which to compare the pharmacophore space covered by each precursor in the set. The transformation of each precursor is carried out using a SMARTS toolkit program, EditSmiles.
EditSmiles is a Smiles and Smarts toolkit program which makes structural changes to a set of molecules. In this context it is used to clip precursors and to add the pseudocore. It is also useful to produce clipped smiles for addition to monomer databases. By iterative addition of various R-groups it may also be used to enumerate combinatorial libraries. EditSmiles either makes changes to a group of atoms or to a bond and defines a rule language to specify these.
An input definition file of rules is provided to the program. The rules are of the following form:
The transformations above would be encoded as
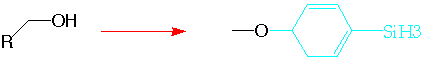
Chg_group [O;H][C;H2] [H] c1ccc([SiH3])cc1 -multi first

Chg_group [N;H](C)NC [H] C[SiH3] -multi firstChg_bond +1 C([SiH3])N [H] NNC[SiH3] [H]
In the second case two rules are needed, the first adds the pseudocore and the second closes the ring to the second nitrogen.
Rules are interpreted into an array of structures containing mol and vector binding objects, together with flag settings.
The following pseudo code is for each input smiles
* note: this functionality comes from combine & du_eliminate in the contrib directory
Potential pharmacophore points are identified and their coordinates calculated using 3D-Features, a expert-system program making use of the Smiles and Smarts toolkits as well as CONCORD.
3 types of pharmacophore point are currently identified by the program.
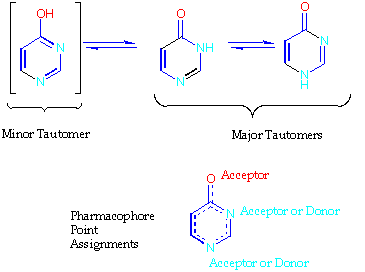
In precursor selection a special "point-of-attachment" type is defined so that all pharmacophore patterns can be oriented relative to that point or set of points. Site points are not currently used in precursor selection. Pharmacophore point assignments for two structures are shown on the first page.
3D-Features - Rule Base
The program is an expert system containing a rule base written in terms of around 300 Smarts targets. These define potential behviours for atoms based on their 2D environments - donor, acceptor, etc. Any atom can hit more than one target. E.g. an atom can potentially be a donor or an acceptor. For example, for the various forms of enolate.
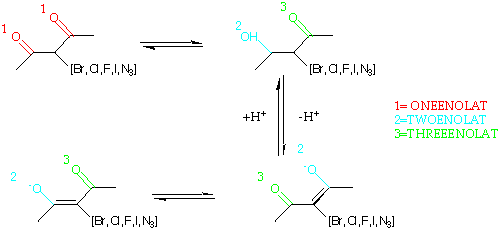
| Group | SMARTS |
|---|---|
| AZIDE | [N]=[N]=N |
| EWG | [Br,Cl,F,I,$AZIDE] |
| ONEENOLAT | O=C([C,c])[C;!H0]([$EWG])C([C,c])=O |
| TWOENOLAT | [O;D1]C([C,c])=C([$EWG])C([C,c])=O |
| THREEENOLAT | O=C([C,c])C([$EWG])=C([C,c])[O;D1] |
| ENOLAT | [$ONEENOLAT,$TWOENOLAT,$THREEENOLAT] |
| ONEG | [$ENOLAT,$ACXM,$ACIDO,$DBOSO,$OM,$OXAM;!$ODNEG] |
| NEG | [$ONEG,$SNEG,$TRZLN,$Arsfnmd] |
3D-Features encodes behaviour on the basis of all major tautomers and all likely ionization states, irrespective of the tautomer or state of the input smiles.
For example, the rule base has rules defining the nitrogen in these two environments to be equivalent for the purposes of charge interaction
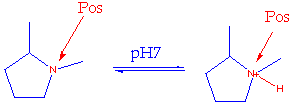
One set of rules will specify that the following three oxygen environments are equivalent and are acceptors only. Other rules will specify that the three environments of each nitrogen are equivalent and are potentially both donor and acceptor.
3D-Features - Program
The program flow is shown in the diagram on the following page. The Smarts targets are loaded and stored as vector bindings. Only the final target bindings $HBD, $HBA, $NEG etc are carried forward to the remainder of the program. Each input smiles is then searched against each target in turn; the results are recorded in a separate pathset for each. The searches are

Structures are first grouped so that all group members have the same set of features, e.g. 2 donors, 1 acceptor, 1 negative + point of attachment. These groups are then subdivided by using each structure as the query to a whole structure 3D search, allowing a tolerance on the match of corresponding distances. A typical tolerance is +/- 0.5Å.
For example the structures shown in the diagram on the following page, taken from the primary alcohol set, all have only one hydrogen bond acceptor in addition to the point of attachment (the pseudocore is shown in green).

Superimposed, the structures are as follows:
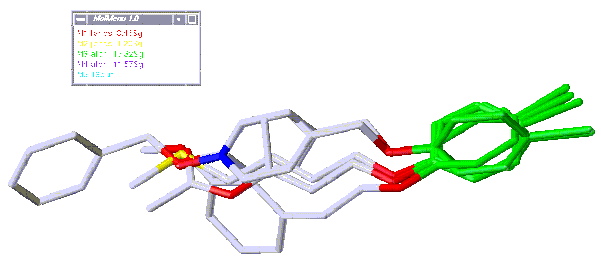
Non-overlapping Families
Each structure heads its own family, and some structures appear in many families. This provides too much information to allow the chemists to make sensible selections from the families. The following simple, and non-optimal, procedure reduces the families so that no structure appears more than once.
Supplier and Price Information
Supplier, quantity and cost information are obtained for each precursor from ACD ORACLE tables. The best price per gram/ml for the minimum required quantity is recorded, together with the relevant supplier, for display when the structures are being selected.
Remove Core/Pseudocore
EditSmiles is run in reverse to remove the (pseudo)core. The original smiles cannot be used as this does not have the feature points marked.
Browsing and Selection
A slightly modified version of the cluster viewer from the Daylight contrib directory is used to browse and select the structures. Price and supplier information is displayed and can be taken into consideration when making selections.
Inventory and ordering of precursors is handled using an ISIS application. Structures are stored in MACCS; lot, container and dispersal information in ORACLE. Ordering will be automated since the existing stockroom system is based on the ORACLE ACD tables.

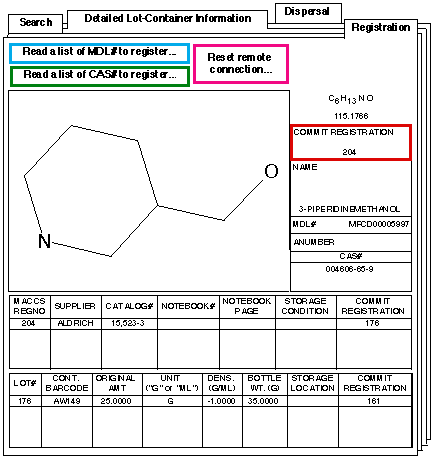
The screenshots show the summary/searching screen and the structure/lot/container registration screens respectively.
Robert Brown,(brownr@abbott.com), Abbott Laboratories, Feb 1996.