The Catalpa
The catalpa's white week is ending there
in its corner of my yard. It has its arms full
of its own flowering now, but the least air
spins off a petal and a breeze lets fall
whole coronations. There is not much more
of what this is. Is every gladness quick?
That tree's a nuisance, really. Long before
the summer's out, its beans, long as a stick,
will start to shed, And every year one limb
cracks without falling off and hangs there dead
till I get up and risk my neck to trim
what it knows how to lose but not to shed.
I keep it only for this one white pass.
The end of June's its garden; July, its Fall;
all else, the world remembering what it was
in the seven days of its visible miracle.
What should I keep if averages were all?
John Ciardi
1. Predict specific ligand-protein complexes, generally.
Apply all we know about intermolecular interactions to understand and predict specifically will happen when a arbitrary potential ligand meets an arbitrary protein. This is "trying to read the mind of God" and is one of the more noble enterprises in chemistry. Unfortunately, it is also one of the most difficult. Evidence suggests that we can explain only a few percent of the observed variance in binding energy. One indication of success is the correct, unsupervised prediction of observed complexes (though this is of little or no actual utility).
This is a much more modest goal and more practical. If one can use what we do know to predict that, e.g., 50% of small molecules in given set can not possibly bind to a given protein, then one has the opportunity to experiment twice as efficiently, by halving (or doubling) the number of effective trials. An indication of success is a prediction threshold below which 100% of known actives are observed among a much larger number of known inactives, i.e., the whole docking process is evaluated as a pre-experimental filter. DockIt was designed primarily for this purpose.
Predict a small number of actives in a large given class of synthesizable structures. At first glance, this seems to be similar to the above problems, but it is very different from statistical and practical points-of-view. Assuming that computation is much less expensive than experiment, for this problem we should be looking at the extreme tail of the prediction. The quality of the average prediction and the ability to simulate known complexes are both quite irrelevant. What is most relevant is how good the predictions are on the extreme tail. E.g., dock a million compounds, pick the 1% which are predicted to bind best, synthesize and test those 10,000 ... what matters is how many of those are active. Indication of success is a high enrichment factor in simulated experiments with measured data. Evidence presented here indicates that DockIt works very well in the tail (much to our surprise).
Although this method might be suitable for predicting average behavior, it is unsuitable for studying the enrichment in extreme tail distribution. This approach is extremely sensitive to decoy assigment errors, i.e., even it the assumption of decoy inactivity is mostly correct, a small number of "false controls" has a huge effect on enrichment analysis results.
To illustate this, assume we do a simulated experiment with 10 known actives and 80,000 decoys, of which 99% (79,200) are actually inactive and 1% (800) are "secretly" active. Let's assume that we have a perfect predictor, so when ranked by score, all actives appear first. What do we find if we select a small tail, say 100, structures? Since our hypothetical prediction function is perfect, all 100 will be active. But we'd expect only (100 * 10 / 810) = 1.2 ~ 1 seeded active and (100 * 800 / 810) = 98.8 ~ 99 to be false negatives which will be counted as mis-predictions. Calculated enrichment in this case will be 1 (found) / (10*100/80000) expected at random = 80x ... pretty good, but this is for a perfect predictor which should be showing 8000x enrichment in this situation. I.e., a 1% error in decoy assignment leads to a 100x error in enrichment calculation.
This situation is difficult to avoid in a sensible manner. We *don't* want to use very diverse structures such as out of ACD, since a large proportion of such structures are reagents which have no chance of binding (it's too easy to pick actives out of such a set, though that might be OK for "filtering" usage, #2 above). Ideally, all molecules tested should be as drug-like as possible, they should all have a good chance of binding, and we'll be testing specific predictions. So it's tempting to use a set of actual drugs or drug candidates such as WDI for decoys, but such data sets contain many somewhat-active drug analogs for each drug, which become false-negatives.
The answer to that question is: very hard. The negative data itself is not valuable, but virtually all companies which generate such data consider the structures to be very proprietary. The fact that we wanted to be able to publish the results if it worked out complicated matters further. We spent several weeks doing a systematic canvas of our drug company friends, with no success. Even structures from long-dead projects (or long-dead companies) are considered holy.
After a few weeks, we discovered that Lutz Weber at Morphochem had prepared a data set specifically for such methods testing. We contacted him and arranged to use it with the understanding that (if things worked out well) we could jointly publish the results in a year's time. What a break.
This data set is composed of 15,840 small molecules from a 3-component Ugi-like reaction scheme, tested against 5 clotting-cascade proteases: factor 10a, tryptase, trypsin, chymotrypsin and UPA (urokinase-type plasminogen activator). It is a marvelously designed data set for the purposes of testing predictions. Positive robotically-done measurements were repeated to assure quality. The proteins are kind of tough, but the ligands are even tougher, that is to say, more wonderful. All 15,830 small molecules look like they'd fit and do pretty well ... they all meet established pharmacophore requirements ... but only a few actually bind well to these enzymes.
One glitch is that 660 of the structures contain Si as a trimethylsilyl substituent (from a trimethylsilyl-methyl-isocyanide reagent). DockIt is not parameterized for silicon so we eliminated these structures for most analyses. Modeling Si as C didn't work particualarly well. DockIt was able to process other 15,180 ligands against all five proteins.
For this experiment, the orders used were docking score ranks, allowing us to evaluate absolute performance and also to compare the performance of different scoring functions. In most of these cases, the data were binned into 1% bins (151 molecules per bin).
To do such an enrichment analysis, one must also define what "active" means. The Morphochem data set has accurately measured activities from about 100uM to sub-micromolar, so we repeated the analysis with "active" defined as 1, 2, 5, 10, 20, 50, and 100 uM. (This was a fortuitous decision.)
For a given protein and ligand set, the main variables are the choice of the scoring function used to drive docking during minimization and which is used for the ranking. These can be same or different (in which case the docked conformations are rescored). For this experiment, we had three scoring functions available, DOCK, PMF and PLP. Of these, only DOCK and PLP are derivitizable and thus fast enough to minimize against.
DockIt does a good job of automatically setting up such runs. Although it's possible to override automatic settings, the results presented here are for DockIt running completely unsupervised.
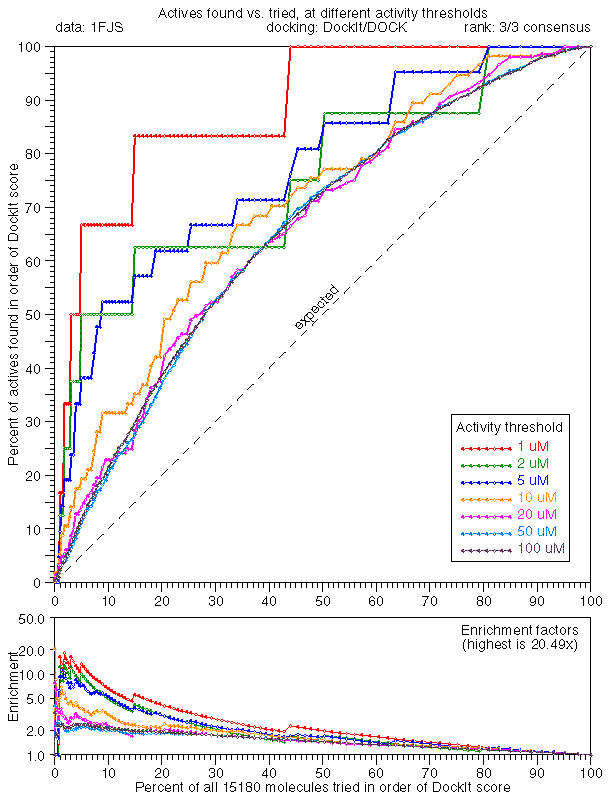
Wow, that's encouraging, 25.0x enrichment in the extreme tail. Note that the 7 colored curves represent different activity definitions, i.e., red <= 1 uM, green <= 2 um, etc. The main plot is percent tried in scoring order vs. percent of actives found. The lower "Enrichment factors" plot is the ratio, on a log scale.
The DOCK-minimized results were rescored by PLP (16.6x), PMF (5.6x), and
Minimizing against PLP produced similar results, with a few surprises. We had great hopes for both minimizing and ranking against PLP (1FJS plp/plp, 8.3x) and it worked pretty well but not nearly as well as rescoring the same docking with the DOCK function (1FJS plp/dock, 16.7x). This seems to be a nearly universal characteristic, at least for this data set.
Rescoring against PMF also produced fair but not great results (1FJS plp/pmf, 8.3x), Which also seems to be a characteristic for this data set.
Consensus scoring was the positive surprise in this run: 3/3 concensus (1FJS plp/33, 115.0x) produced remarkable results. Part of this is due to binning (these are minimum rather than "instantaneous" enrichments, but they don't differ by much).
This seems to be worth looking at more closely. Here are the same results, zoomed 10x horizontally. Looking at the best 3% of the rankings by each score, there are 22 in the concensus set (22/15180 = 0.145%) and 1/6 of the 6 sub-1uM actives have been found. We'd expect to find 6 * 22 / 15180 = .008696 actives at random, so we're doing (at least) 1 / 0.00115 = 115.0x better than random. When 80 appear in the concensus set, we've found 2/6 corresponding to 2 / (6 * 80 / 15180) = 63.3x.
To our amazement, this completely unsupervised run produced 1C5P dock/dockpmf, showing an enrichment of 772.7x! How could that be? The 7th "pick" was one of the three actives. It's probably a anomaly, but what's the chance that?
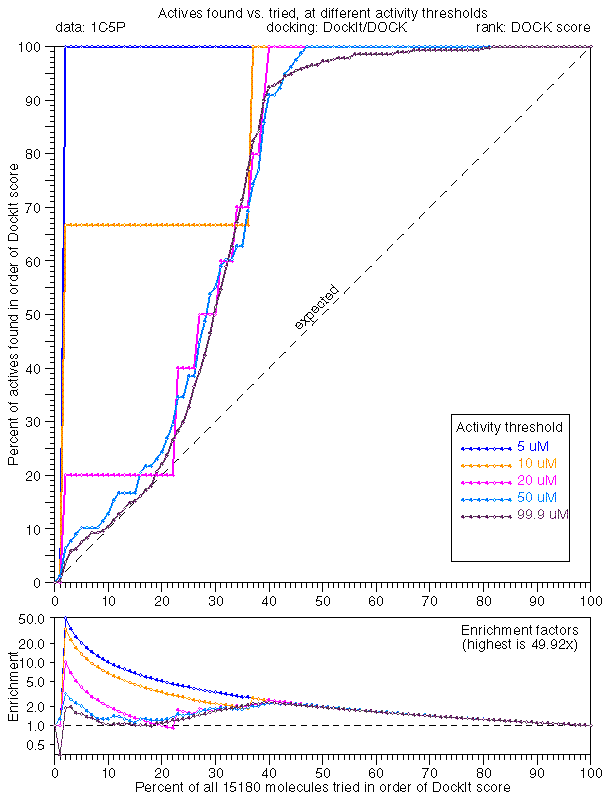
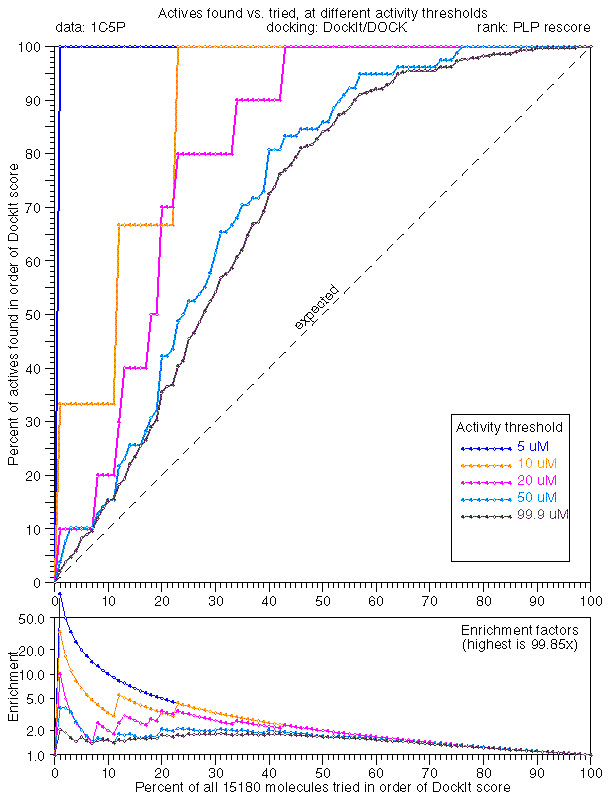
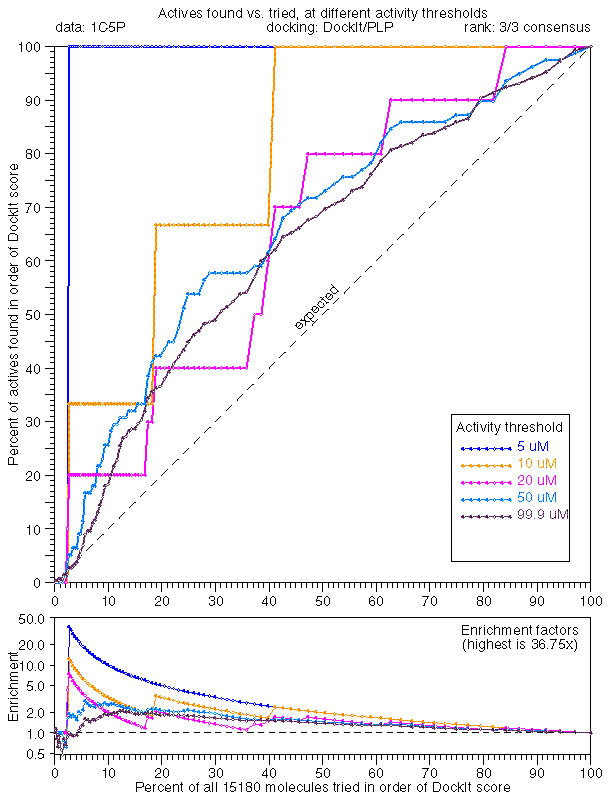
In general, the extreme tail enrichment worked very well for trypsin. 1C5P dock/dock (25.0x), 1C5P dock/plp (99.9x), and 1C5P dock/33 (36.1x) all showed excellent tail enrichment which we had by now came to expect. Given our new high standards, rescoring with PMF didn't seem to work that well (1C5P dock/pmf, 16.6x).
Docking with PLP minimization was comparable when ranked by consensus (1C5P plp/33, 36.8x) and PLP score (1C5P plp/plp, 33.3x). Rescoring PLP dockings with DOCK (1C5P plp/dock, 6.7x) or PMF (1C5P plp/pmf, 5.8x) showed poor performance in tail enrichment.
2CHA (chymotripsin) is the most "different" enzyme in this set, with respect to the nature of it's hydrophobic-loving binding site. Only two very active ligands were observed (0.09 uM) then a bunch of moderate containing Si (which we couldn't model). So the data is kind of sparse. Even so, PLP scorings found one of the two actives in the first 1% bin, whether minimized against DOCK (2CHA dock/plp, 25.0x) or PLP (2CHA plp/plp, 25.0x).
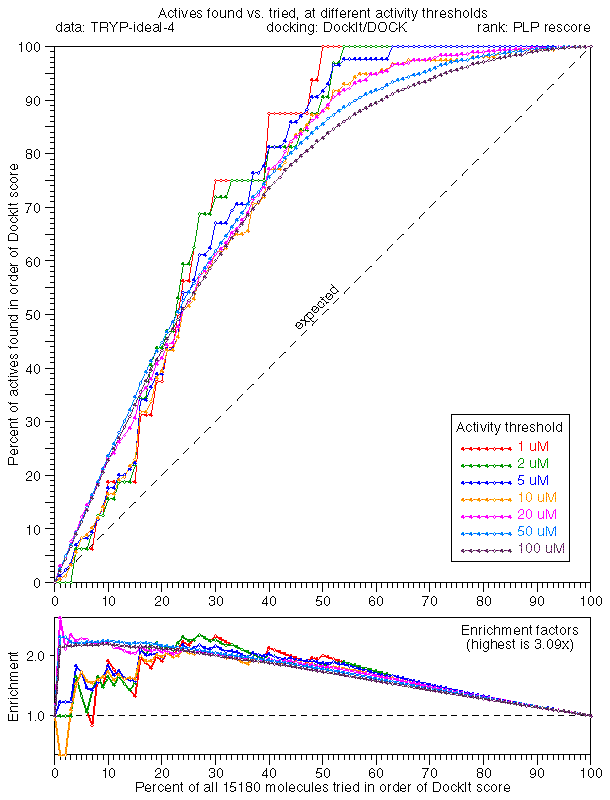
The TRYP (tryptase) tetramer structure which we docked against is a Morphochem structure which seems nearly identical (at least in sequence) to PDB's 1A0L. The data for this structure do not seem comparable to the others. It contains a huge number of binders (~30% of the total), which might be right or we might have screwed something up in the data preparation for this enzyme's data. We'll have to go back and check everything. Enrichment plots show fair filtration performance with DOCK (TRYP dock/dock) and (TRYP dock/plp) rankings, but little or no tail enrichment. Ranking by PMF score (TRYP dock/pmf) is worse than random. Again, we're not too sure we did this one correctly, but we're reporting it as a matter of completeness.
Each run was also done in random order, which shouldn't show good enrichment, and gratifyingly, doesn't.
Each run was also done in original order (appears to be sorted by robot programming code) which isn't random, doesn't actually prove anything, but still looks gratifyingly terrible.
A limited number of runs were done switching observed data and predicted order between enzymes. I.e., using the observed data for 1FJS binding with the ranking from 1C5P docking (two very similar enzymes), we see that generic binding predictions are retained but specific binding predictions on the tail are completely lost. These plots for 3/3 consensus, dock/pmf consensus, and PLP scoring are typical.
The TRYP/1C5Z switch shows similar effect: dock/dock, dock/plp.
| 1FJS | dock | 33 | xx |
| 1FJS | dock | dock | xx |
| 1FJS | dock | plp | xx |
| 1FJS | dock | pmf | xx |
| 1FJS | plp | 33 | xx |
| 1FJS | plp | 33 | xx |
| 1FJS | plp | dock | xx |
| 1FJS | plp | plp | xx |
| 1FJS | plp | pmf | xx |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}