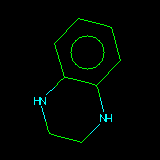
| C1CNc2ccccc2N1 | Alignment starts by defining the substructure which each structure has in common. This is done by reading a SMARTS string supplied by the user. |
|
The first structure is read and the coordinates of the first three atoms matching the SMARTS are retained for later reference. Call these coordinates A1, B1 and C1. The structure is output unchanged. |
|
As each subsequent structure is read, the coordinates of the first two atoms (An and Bn) matching the SMARTS are used to compute the angle between the lines(bonds) A1B1 and AnBn. |
|
All atoms of structure #n are rotated around the z-axis by the negative of that angle. All atoms of structure #n are translated in x and y so that B1 superimposes with Bn. The coordinates C1 are compared to the rotated coordinates Cn. If necessary, the coordinates of all atoms of structure #n are rotated/flipped by 180 degrees around AnBn. |
The following other options are also available: